Mecanismos bioquímicos desencadenados después de la lesión de médula espinal
- Inicio
- Comité Editorial
- Lineamientos
- Carta de Cesión de Derechos
- Información Legal
- Acerca de la Revista
- Bases de Datos
- Contacto
- ISSN 2007-3054
- Centro de Investigaciones Cerebrales
Universidad Veracruzana
Sandra Yasbeth Lara-Aparicio1, Ángel de Jesús Laureani-Fierro1, Luis Isauro García-Hernández2, Ricardo Ortiz-Pulido3, Consuelo Morgado-Valle2, Luis Beltrán-Parrazal2, María Elena Aguilar-Hernández2, Jorge Manzo-Denes2, César Antonio Pérez-Estudillo*2
1Doctorado en Investigaciones Cerebrales. Universidad Veracruzana.
2Centro de Investigaciones Cerebrales. Universidad Veracruzana Xalapa.
3Dirección de Actividades Deportivas de la Universidad Veracruzana.
Resumen
Abstract
Introducción
Incidencia de la lesión de la médula espinal
Mecanismos bioquímicos desencadenados después de la lesión de médula espinal
Fase aguda
Fase subaguda
Fase crónica
Conclusiones
Conflicto de intereses
Agradecimientos
Referencias
Mail
Resumen: La médula espinal constituye parte del Sistema Nervioso Central junto con el cerebro, el cerebelo y el tallo cerebral; siendo un puente de información sensorial, motora y autonómica entre las estructuras supraespinales y la periferia. La médula espinal es considerada como la vía de comunicación entre los núcleos encefálicos con los demás órganos del cuerpo, por lo que una lesión (parcial o completa) en esta estructura produce alteraciones anatómicas y funcionales. La lesión de la médula espinal (LME) resulta en una patología compleja dividida en tres fases: a) el choque espinal, donde pierde funciones de manera temporal o permanente, dependiendo el grado de la lesión; b) el daño primario (fases aguda y subaguda), donde inician los primeros procesos bioquímicos como lesiones vasculares, choque neurogénico, liberación y sobreproducción de radicales libres, peroxidación lipídica, excitotoxicidad, etc., que generan una muerte neuronal primaria; y c) el daño secundario (fase crónica), que da paso a la generación de otros mecanismos bioquímicos como procesos inflamatorios, pérdida de mielina y de crecimiento axonal, degeneración walleriana, cicatriz glial, etc.) que culminan en una segunda muerte neuronal. El conocimiento incompleto de los cambios bioquímicos, fisiológicos y neuroanatómicos en el daño primario y secundario que ocurren en diferentes tiempos después de la lesión espinal ha generado que los tratamientos actuales disponibles sean parcialmente exitosos. Esta revisión comprende una recopilación de diversas investigaciones que abarcan todos los procesos bioquímicos involucrados en la LME que se conocen actualmente.
Palabras clave: lesión de médula espinal, daño primario y secundario, muerte celular.
The spinal cord constitutes part of the Central Nervous System along with the brain, cerebellum, and brainstem; being a bridge of sensory, motor, and autonomic information between the supraspinal structures and the periphery. The spinal cord is considered as the communication pathway between the encephalic nuclei with the other organs of the body, so a lesion (partial or complete) in this structure produces anatomical and functional alterations. Spinal cord injury (SCI) results in a complex pathology divided into three phases: a) spinal shock, where it loses functions temporarily or permanently, depending on the degree of the injury; b) primary damage (acute and subacute phases), where the first biochemical processes such as vascular lesions, neurogenic shock, release and overproduction of free radicals, lipid peroxidation, excitotoxicity etc., which generate primary neuronal death, begin; and c) secondary damage (chronic phase), which leads to the generation of other biochemical mechanisms such as inflammatory processes, loss of myelin and axonal growth, Wallerian degeneration, glial scar, etc.) culminating in a second neuronal death. Incomplete knowledge of the biochemical, physiological, and neuroanatomical changes in primary and secondary damage that occur at different times after spinal injury has resulted in the currently available treatments being partially successful. This review comprises a compilation of various investigations covering all the biochemical processes involved in SCI that are currently known.
Keywords: spinal cord injury, primary and secondary damage, cell death.
La médula espinal (ME) constituye el primer nivel integrador del Sistema Nervioso Central (SNC) transmitiendo información proveniente del cerebro al Sistema Nervioso Periférico (SNP), por lo que la lesión de la médula espinal (LME) frecuentemente conlleva a la pérdida de control supraespinal de funciones motoras, sensoriales y autonómicas por debajo del sitio afectado, asociada a una serie de procesos bioquímicos que se desencadenan.1,2 Lo primero que se presenta es el choque espinal que puede durar desde unos minutos, unas horas, unos días o unos meses procediendo del grado de la lesión y dependiendo de la especie. Posteriormente, se desencadena el daño primario que involucra a todos los procesos bioquímicos que se llevan a cabo durante las primeras 24 horas. Todos estos procesos bioquímicos generan otros que pertenecen al daño secundario y que corresponden a las 24 horas, días semanas y/o meses subsecuentes al daño espinal.3 De esta forma, la LME produce una discapacidad crónica muy importante que afecta a millones de personas en todo el mundo cada año (aproximadamente 250-500 mil personas)4 y aunque actualmente existen tratamientos neuroprotectores para restaurar la LME, esta recuperación funcional ha sido parcial y no total debido a que aún se desconocen todos los mecanismos bioquímicos y circuitos neuronales involucrados en esta fisiopatología.
2. Incidencia de la lesión de la médula espinal
La LME afecta biopsicosocialmente a las personas que las sufren sin importar el género, la edad, el estatus social, la nacionalidad, etc., quienes regularmente se encuentran en etapas productivas y, en la mayoría de las veces, se convierten en una “carga” para sí mismos, para sus familias y para la sociedad.5,6 La Organización Mundial de la Salud (OMS) estableció en el 2013 que entre 250-500 mil personas sufren una LME cada año en todo el mundo4 y según la National Spinal Cord Injury Statistical Center, en Estados Unidos, cada año se presentan 12,500 casos nuevos de LME.7 En México, en el año 2007, se calculó que anualmente esta lesión se presenta en 18.1 personas por millón de habitantes, siendo más frecuente en hombres en edad productiva (16 a 35 años de edad). 8 En el año 2017, la clínica de lesión medular en el Centro de Rehabilitación e Inclusión Infantil del Estado de México obtuvo un ingreso mensual de 47 pacientes (27 masculinos y 20 femeninos) en un rango de edad de entre 9 meses y 19 años. 9
Por otro lado, se sabe que hasta el 45% de las LME resultan en anormalidades completas (pérdida total de sensación y función debajo del nivel de la lesión) y 55% a incompletas (pérdida parcial de la función); aumentando el riesgo de morir prematuramente aproximadamente 2-5 veces en comparación con personas sin lesión debido a la serie de procesos dañinos que se desencadenan y de los que se hablará más adelante, y más si los pacientes residen en países de ingresos bajos y medios.8,10 En el Estado de México, aproximadamente el 55.3% de los casos reportados son de LME incompletas y el 44.6% son completas.9 Aunque las causas más frecuentes de la lesión medular (principalmente a nivel cervical caudal o inferior, unión cérvico-torácica y unión dorso-lumbar) son ocasionadas por causas prevenibles como los accidentes de tránsito, laborales o deportivos (en más del 50% de los casos), 4,6 entre el 25-60% de los casos restantes pueden estar relacionados con lesiones por arma de fuego, caídas de varios metros de altura, lesiones por ondas explosivas, clavados en aguas poco profundas, etc.3,6
3. Mecanismos bioquímicos desencadenados después de la lesión de médula espinal
Después de una LME, dependiendo del nivel de la misma e independientemente de su causa, se desencadenan una serie de procesos autodestructivos que pueden ser mecanismos separados aunque relacionados entre sí: el choque espinal que comprende el daño mecánico inicial, los procesos del daño primario que conciernen a los cambios bioquímicos iniciales derivados del impacto que se dan en las primeras horas del daño, que abarca de 0-24 horas; y los procesos del daño secundario corresponden a los cambios bioquímicos posteriores debido a los efectos vasculares y bioquímicos que surgen a partir de las 24 horas posteriores.3 Cada uno de estos tres mecanismos desencadena una serie de respuestas bioquímicas que se dividen en diferentes etapas, según el autor que los refiera, y siguen una secuencia temporal particular pero superpuesta. El rango de tiempo en que dura cada fase también varía con respecto a los autores (Tabla 1, partes 1-3). Para fines de esta revisión, se manejarán tres fases: aguda (Figura 1), subaguda (Figura 2) y crónica (Figura 3).
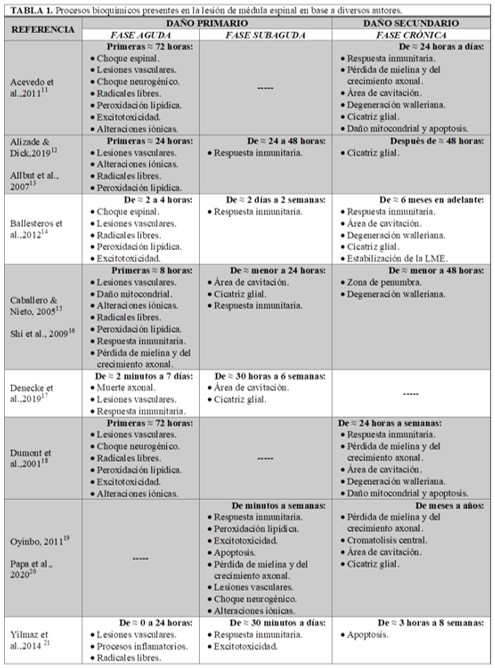
Tabla 1. ≈: aproximadamente; LME: lesión de médula espinal.
Es importante mencionar que los procesos bioquímicos desencadenados tras la LME (en áreas rostrales) no sólo se observan en el mismo tejido espinal, sino que pueden afectar a otras regiones distales y en diferentes tiempos tal como se ha reportado en el área lumbosacra (una zona más caudal) de la médula espinal,22 en algunos núcleos hipotalámicos (núcleos paraventricular y supraóptico),23 en diversos núcleos del tallo cerebral (formación reticular, área postrema y tracto solitario)24 y también en el cerebelo.25,26
El primer trauma después de la LME es conocido como choque espinal, que produce una pérdida temporal de los procesos sensoriales, motores y autonómicos de la sustancia gris central (debido a que es más blanda y vascularizada que la sustancia blanca). El choque espinal en humanos consta de cuatro fases: la fase 1 abarca del día 0 al día 1 y se caracteriza por la pérdida de información descendente con una disminución de las respuestas reflejas (hiporreflexia) y una ausencia de reflejos inconscientes tras estímulos externos (arreflexia). La fase 2 abarca del día 1 al día 3 y se caracteriza por el retorno de los reflejos y una hipersensibilidad; la fase 3 comprende de la semana 1 a la semana 4 donde se observa una hiperreflexia inicial, así como un incremento de la sinapsis. Por último, la fase 4 inicia del mes 1 al 12vo mes en donde culmina la hiperreflexia de la fase anterior y muestra un crecimiento de sinapsis basada en los somas.6,11,27 Esta pérdida funcional, a su vez, generará alteraciones morfológicas que no se presentarán por separado sino que se encontrarán relacionadas, generando el daño primario, que es inmediato e irreversible y desencadena una variedad de procesos bioquímicos que aumentan o disminuyen la gravedad de la patología sobre el daño secundario.23
Existen adicionalmente cuatro tipos morfológicos de choque espinal: el impacto con compresión persistente (que es el mecanismo más común), consiste en presiones permanentes de la médula espinal después de un impacto; por ejemplo, en las fracturas por estallido con fragmentos óseos retropulsados que comprimen la médula, dislocaciones de fractura y roturas agudas del disco. Otro tipo de lesión morfológica es el impacto con compresión transitoria, que involucra un impacto con compresiones efímeras como se observa con las lesiones por hiperextensión en individuos con enfermedad degenerativa subyacente de la columna cervical. También pueden producirse lesiones por distracción o estiramiento forzado de la columna en el plano axial debido a la flexión, extensión, rotación o dislocación de la médula espinal, produciendo cizallamiento o estiramiento de esta y/o suministro de sangre. El cuarto tipo morfológico de choque espinal es la laceración o transección, que consiste en la disrupción o desgarro de la médula espinal por proyectiles, dislocación aguda de fragmentos óseos o estiramientos graves. Puede ocurrir en diversos grados, desde lesiones menores hasta la sección completa transversal; siendo sus principales mecanismos: el impacto más la compresión persistente, un solo impacto con una sola compresión transitoria, la distracción y la laceración/transección.11,18,19
4.2. Lesiones vasculares y procesos inflamatorios en la médula espinal
Tras el choque espinal, se produce un daño en la materia gris y un marcado incremento en la permeabilidad de la barrera hematoencefálica (BHE)14,18 y la microcirculación, especialmente los vasos sanguíneos pequeños (vénulas y capilares), que se daña produciendo pequeñas áreas de hemorragias o petequias que alteran el flujo sanguíneo11,18 (hipoperfusión que puede durar días o incluso meses)19,21 , generando un estado de hipoxia, isquemia (que empeora progresivamente durante las primeras horas), 11,19 anoxia e hipoglucemia, 15 que producen infartos locales.11 Debido a que la presión de perfusión está directamente conectada a la presión arterial sistémica, el daño a la médula espinal se exacerba.21 La permeabilidad de la membrana celular comprometida también se asocia con la activación de las proteasas.19 Esta alteración en la circulación sanguínea se presenta en forma de edema (donde las células sanguíneas y las proteínas del suero invaden el área lesionada) y de hipoperfusión [generado por los tromboxanos, los leucotrienos, el factor activador de plaquetas (PAF), la 5-hidroxitriptamina (5-HT o serotonina) y los opioides endógenos11,15 (siendo la dinorfina la más afectada en la LME humana)].21 Las neuronas que se encuentran o pasan en el nivel de la lesión están físicamente alteradas y muestran una disminución en el espesor de la mielina y en la transmisión nerviosa.11,18
Los cambios en el flujo sanguíneo afectan principalmente a la sustancia gris,15 ya que ahí se encuentran las neuronas que son de alta demanda metabólica y tienen una densidad cinco veces mayor de lechos capilares que la sustancia blanca,12 y los cambios son irreversibles dentro de la primera hora posterior al trauma 11,19 que inducen la muerte neuronal primaria.15,21 La sustancia blanca es más resistente a los efectos de la isquemia e hipoxia, 15 dañándose de forma irreversible dentro de las primeras 72 horas después de la LME.11,19 El flujo sanguíneo de la sustancia blanca generalmente vuelve a los niveles normales dentro de los 15 minutos posteriores a la lesión; mientras que hay múltiples hemorragias en la materia gris12,19 que dan como resultado una etapa de perfusión reducida que puede preceder a un período de hiperemia que se origina, posiblemente, por una reducción del pH perivascular por acumulación de metabolitos ácidos como el lactato; exacerbando la lesión y la muerte celular.11,18 La isquemia inicia la producción de radicales libres perjudiciales y otros productos tóxicos (como radicales libres y peroxidación lipídica), con un efecto más pronunciado durante el período temprano de reperfusión;14,18,19 ocasiona un transporte alterado de glucosa y oxígeno a los tejidos, resultando en una disminución de las reservas de trifosfato de adenosina (ATP). La disminución del pH del tejido debido a la acidosis láctica, la congestión y la estasis venosa debido a la acumulación de fibrina y plaquetas, el daño endotelial capilar, el edema, las hemorragias petequiales y las citocinas vasoactivas son algunas de las causas de la isquemia.21
La muerte celular de neuronas vecinas produce cambios en la permeabilidad de la membrana de la mitocondria en las neuronas intactas, reduciendo su potencial de membrana, que a su vez genera la disminución del rango metabólico y de las concentraciones de fosfatos de alta energía, y contribuyendo a la hinchazón osmótica, a la lisis mitocondrial11,15 y a la predominancia del ambiente medular anaeróbico (dentro de las primeras 4 horas; promovido también por la isquemia).15,21 Este evento produce una excesiva acumulación de calcio (Ca2+) en la mitocondria; proceso que, junto con el estrés mecánico, las reacciones inflamatorias y la alteración de la transducción de señales tróficas contribuyen al daño mitocondrial. Adicionalmente, la permeabilidad aumentada de la membrana mitocondrial externa a las proteínas apoptogénicas facilita su liberación al citosol y representa un mecanismo clave en la inducción de la apoptosis y la muerte neuronal.11 Después de 4 a 24 horas, el rango metabólico tiende a aumentar y cambia a un metabolismo oxidativo en el tejido que permanece intacto, originando acidosis láctica.15
4.3. Muerte axonal en la médula espinal
Los axones mueren a través de cuatro procesos diferentes según el tipo de lesión: pérdida de axón inducida por poros, degeneración axonal aguda, retracción axonal lenta y degeneración walleriana: a) La pérdida de axón inducida por poros se da en LME no transeccionadas, sino contusivas, creando poros en los axones afectados, a través de los cuales el calcio puede ingresar inmediatamente. El aumento resultante en el Ca++ intra-axonal coincide con la inflamación axonal, iniciada principalmente en los primeros 30 minutos, y es un predictor de fragmentación axonal mas no siempre conduce a él. Si los mecanoporos continúan interrumpiendo una membrana axonal después de la contusión, es cada vez más probable que el axón se fragmente; b) La degeneración axonal aguda dura menos de 5 minutos y ocurre en un intervalo de tiempo de 2 minutos a 4 horas después de la lesión; es un proceso relativamente rápido que conduce a la fragmentación de los componentes del axón proximal y distal (en relación con el sitio de la lesión) simultáneamente, fragmentándose en direcciones opuestas a la lesión (retrógrada y anterógradamente); c) La retracción axonal lenta también se observa en los componentes del axón proximal y distal pero a tasas más lentas y por retracción en lugar de fragmentación. Es un proceso "limpio" que retrae todo el extremo del axón (a diferencia de los procesos de fragmentación) horas después de la lesión, induciendo la formación de grandes estructuras bulbosas en la mayoría de los tocones de axón cortados, denominados bulbos de retracción; d) La degeneración walleriana que se explicará más adelante es un proceso de fragmentación que deja trozos de axones durante horas después de la lesión (cuya fase final se da a las 30 h y hasta las 6 semanas); es decir, conduce a la degeneración de tramos muy largos de axones, con una tasa de degeneración similar a la degeneración axonal aguda; sin embargo, difiere de ésta última al fragmentar el extremo distal y no proximal del axón. Comienza en el extremo lesionado del segmento distal del axón, lo que lleva a un adelgazamiento de todo el axón, progresando distalmente a lo largo del axón. Curiosamente, la degeneración walleriana no se limita a los axones cortados claramente identificados, sino que también puede ocurrir en axones aparentemente no lesionados donde se inicia cerca de la lesión.17
4.4. Radicales libres y peroxidación lipídica en la médula espinal
El origen de los radicales libres o especies reactivas de oxígeno15,28,29 (ROS;19 como superóxido, radicales en hidroxilo y el óxido nítrico) después de una LME se puede deber a cuatro mecanismos:11,28 a la isquemia-reperfusión,21 a la actividad de los macrófagos sanguíneos que atraviesan la BHE destruida, a la muerte eritrocitaria en el sitio de lesión que provee una fuente de hierro que cataliza la formación de más radicales libres de oxígeno y al incremento de Ca++ intracelular estimulado por la liberación de aminoácidos excitatorios.28 Por su parte, los otros oxidantes de alta energía (como el peroxinitrito) se producen durante la isquemia con un marcado aumento durante el período de reperfusión temprana.11 La excesiva producción de ROS excede la capacidad celular de protección antioxidante,11,29 ocasionando lisis de la membrana celular, pérdida neuronal y daños particularmente en el citoesqueleto y los orgánulos, así como un desbalance conocido como estrés oxidativo,19,29 que contribuye al daño secundario del trauma generado a través de múltiples vías celulares (que regulan a su vez la formación de compuestos de nitrógeno y de oxígeno altamente reactivos) que incluyen las óxido nítrico sintasas11,18 (iNOS),19 activación de fosfolipasas mediada por calcio, xantina oxidasa, activación de células inflamatorias y las reacciones de Fenton y Haber-Weiss.11,18 El estrés oxidativo provoca la oxidación de proteínas, lípidos y ácidos nucleicos mediante la inactivación de las enzimas de la cadena respiratoria mitocondrial como el gliceraldehído 3-fosfato deshidrogenesa (GAPDH), la inhibición de la sodio-potasio ATPasa y la inactivación de los canales de sodio (Na+)11,15,29 y otras perturbaciones oxidativas de proteínas clave. El estrés oxidativo comparte puntos en común con otros mecanismos patogénicos que inducen lesiones de la médula espinal como lo son la sobrecarga de Ca++, la liberación de citocromo C mitocondrial, la activación de caspasa, la apoptosis y la excitotoxicidad.18
La oxidación de lípidos o ácidos grasos (peroxidación lipídica) debido a las altas concentraciones de radicales libres,11,14,18 a la disfunción mitocondrial, a la descomposición del ácido araquidónico y a la activación de la iNOS, lesionan las membranas celulares, ocasionan daño oxidativo y nitrativo a proteínas y ácidos nucleicos, produciendo muerte celular asociada a disfunción de los organelos; contribuyendo así con la alteración de la homeostasis de Ca++, es decir, forman un círculo vicioso de procesos que aumentan la muerte celular. En la peroxidación lipídica, los ROS absorben un electrón de una molécula lipídica, que a su vez se vuelve menos estable, desencadenando una reacción en cadena que finalmente conduce a la lisis de la membrana y la muerte por necrosis y apoptosis. Además, el daño oxidativo exacerba la disfunción mitocondrial y contribuye a la sobrecarga de Ca++ intracelular activando las proteasas que provocan la descomposición de las proteínas del citoesqueleto.14,19 Al liberarse y oxidarse los ácidos grasos polinsaturados y el ácido araquidónico, se propicia la formación de araquidonato que, mediante la 5-lipooxigenasa (5-LO), genera leucotrienos; y a través de las ciclooxigenasas (COX), crea prostaglandinas y tromboxanos. El exceso de araquidonato (aproximadamente 5-15 minutos posteriores a la LME) promueve el daño tisular, aumenta la permeabilidad vascular y la inflamación.15,30 También los tromboxanos, prostaglandinas y leucotrienos, por su parte, que coadyuvan en la hipotensión. Los tromboxanos y leucotrienos afectan al flujo sanguíneo y promueven la inflamación. Además, la vasoconstricción provocada por la activación de la fosfolipasa A2 y la agregación de las plaquetas contribuyen a la reducción postraumática del flujo sanguíneo.11,15 Es así como el daño a las membranas celulares y la peroxidación lipídica da como resultado la producción de más ROS, los cuales continúan dañando las membranas, resultando en una mayor peroxidación de lípidos y formación de radicales libres. Este ciclo continúa a menos que sea detenido por antioxidantes endógenos,18 como las vitaminas A, C y E, los flavonoides, los carotenoides, los fenólicos, la acetilcisteína, el selenio exógeno, el zinc, la superóxido dismumatasa, entre otros.31
4.5. Excitotoxicidad en la médula espinal
El daño indirecto producido por los ROS y el nitrógeno al parénquima de la médula espinal, así como los cambios en la microcirculación y por la isquemia conducen a una liberación y acumulación de neurotransmisores excitatorios dentro y alrededor del sitio de la lesión como el glutamato11,19 y aspartato15 que alcanzan los niveles tóxicos entre los 15 a 30 minutos, volviendo a sus niveles basales después de 60 minutos y manteniendo su toxicidad hasta los 120 minutos)12,14,21 y, a su vez, una activación excesiva de sus receptores (proceso conocido como excitotoxicidad).11 La activación de los receptores de glutamato N-metil-D-aspartato (NMDA) y α-amino-3-hidroxi-5-metilisoxazol-4-propionato-kainato (AMPA-kainato) da como resultado una acumulación temprana de Na+ intracelular, produciendo un edema citotóxico posterior y una acidosis intracelular. La falla de la ATPasa sodio-potasio exacerba aún más la acumulación intracelular de Na+ y agua, así como la pérdida extracelular de potasio (K+) que coadyuva a la excitotoxicidad. Por su parte, la acumulación intracelular de Ca++ produce alteraciones profundas en la fisiología y daños posteriores, denominándose la "vía común final de muerte celular tóxica" en el SNC. La excitotoxicidad, particularmente mediada por el receptor NMDA, inicia una compleja cascada de eventos que finalmente da como resultado la génesis de moléculas reactivas18 que contribuyen a empeorar la isquemia, alterar la función microcirculatoria y dañar directamente al tejido de la médula espinal. De manera indirecta y como ciclo repetitivo, la excitotoxicidad genera alteraciones iónicas11,18 que lisan las células y liberan más glutamato, alteraciones en la función microcirculatoria e isquemia secundaria 14,19 y genera más especies reactivas de oxígeno y nitrógeno, las cuales permiten más liberación de este neurotransmisor.18 Es importante mencionar que la excitotoxicidad no sólo afecta/destruye a las neuronas sino también a la glía, específicamente a los oligodendrocitos.12,32
4.6. Alteraciones iónicas y electrolíticas en la médula espinal
La formación del edema generado por el aumento de la presión local comprime el tejido y produce una variación anormal en las concentraciones de electrólitos; además, induce la liberación de bradicininas, citocinas, histaminas y óxido nítrico que, a su vez, contribuyen a aumentar la permeabilidad vascular y la formación de radicales libres.15 Por otra parte, la activación exacerbada de los receptores de glutamato (NMDA y Kainato-AMPA) produce la acumulación rápida de Na+ intracelular con el subsiguiente edema citotóxico y acidosis intracelular. La falla de la ATPasa sodio-potasio empeora la acumulación intracelular de Na+ y agua y la pérdida extracelular de K+.11
Las variaciones leves sobre las concentraciones iónicas de Ca++, Na+ y K+ en el fluido intersticial, perturban la excitabilidad y la transmisión sináptica. Tres mecanismos coadyuvan al incremento intracelular de Ca++: la sobreactivación de los canales dependientes de voltaje o del canal asociado al receptor para glutamato tipo NMDA; su liberación de los sitios de almacenamiento intracelular localizados en la matriz mitocondrial y retículo endoplásmico; y la inhibición de la calcio-ATPasa (que permite la salida de Ca++ de la célula).15 Tras la LME, los canales de gran conductividad o dependiente de voltaje y de calcio (Bk del inglés Big Potassium o Maxi K+), que residen en la región yuxtaparanodal axónica, quedan expuestos debido a que las neuronas pierden la mielina de esa región, generando su sobreactivación y contribuyendo a la función fisiológica alterada de la ME al ser influenciados por los niveles de Ca++ intracelular y al ser extremadamente sensibles a niveles elevados de ROS, inhibiendo su conductancia.33
El Ca++ intracelular comienza a aumentar a partir de los 30 minutos después de la lesión, a los 45 minutos llega a su segundo pico, sube de nuevo 4 horas después, luego a las 8 horas y alcanza otro máximo a las 24 horas, punto en el que el grado de daño es irreversible; permaneciendo elevado al menos durante una semana lo que produce daños en la mitocondria de las neuronas que aún permanecen vivas al alterar su habilidad para llevar a cabo la respiración celular y la fosforilación,11,15 una regulación positiva de óxido nítrico13 y la liberación de citocromo C, iniciando así una secuencia programada alterna de activación de las caspasas. Otros mecanismos dañinos desencadenados por la excesiva concentración interna de este ion son:11 la activación de las proteasas, lipasas y fosfolipasas dependientes de calcio (como calpaínas, fosfolipasa C y A2, lipoxigenasa y ciclooxigenasa) que alteran la membrana celular al hidrolizar los fosfolípidos y liberan los ácidos grasos polinsaturados;15,18 así como cambios en la expresión génica y apoptosis.19 Los ácidos grasos polinsaturados alcanzan su primer pico de acumulación entre los 5 a 15 minutos posteriores a la lesión. Los ácidos grasos liberados coadyuvan a la producción de araquidonato, cuyo metabolismo produce tromboxanos, leucotrienos y ROS, promoviendo el daño tisular e inflamación.15 Un modo particularmente poderoso de entrada de Ca++ dentro de los axones lesionados en la sustancia blanca implica una fuga inicial de Na+ debido a la lesión aguda, lo que impulsa al intercambiador de sodio-calcio a importar niveles dañinos de Ca++.11
Las calpaínas degradan componentes estructurales importantes en el SNC, incluidas las proteínas estructurales del complejo axón y mielina. Además, otras proteasas y quinasas dependientes de calcio destruyen las membranas celulares y provocan la disolución de ciertos componentes de la ultraestructura celular, como los neurofilamentos. La activación de la lipasa, la lipoxigenasa y la ciclooxigenasa-1 (COX-1) en microglía y macrófagos da como resultado la conversión del ácido araquidónico en tromboxanos, prostaglandinas y leucotrienos produciéndose a niveles elevados en cuestión de minutos. El aumento del ácido araquidónico se da aproximadamente a las 24 horas después de la lesión y se asocia con la inhibición de la ATPasa sodio-potasio y el edema tisular, lo que contribuye a la reducción del flujo sanguíneo al causar agregación plaquetaria y vasoconstricción, a la respuesta inflamatoria y a la peroxidación lipídica.18
Otros iones que también muestran un desequilibrio en sus concentraciones tras la LME son el K+ y el magnesio (Mg++). El incremento en la concentración extracelular de K+ promueve la despolarización excesiva de las neuronas, afectando negativamente la conducción neuronal y siendo el posible factor causal crítico subyacente al choque espinal. El agotamiento o disminución intracelular de Mg++ puede tener efectos nocivos en los procesos metabólicos, como la glucólisis, la fosforilación oxidativa y la síntesis de proteínas, además de afectar negativamente ciertas reacciones enzimáticas en las que este ion sirve como cofactor. También puede contribuir aún más a la acumulación intracelular de Ca++ y a los procesos fisiopatológicos asociados ya descritos. Los niveles de ciertos neurotransmisores y péptidos también cambian después de la lesión, por ejemplo, el aumento de los opioides endógenos contribuye a la activación de los receptores opioides μ y δ exacerbando, los cuales exacerban las disminuciones en el flujo sanguíneo y promueven el proceso excitotóxico.18 La concentración de acetilcolina y 5-HT también aumentan, causando vasoconstricción y promoviendo la activación plaquetaria y la permeabilidad endotelial.18,34
5.1. Respuesta inmunitaria en la médula espinal
La muerte eritrocitaria en el sitio de lesión a raíz de las hemorragias libera productos de la hemoglobina que son fuertes quimioatrayentes de los leucocitos, activando así al sistema inmunitario11,15 -leucocitos-18 de manera bifásica: primero se produce la infiltración/reclutamiento de granulocitos polimorfonucleares (neutrófilos o leucocitos polimorfonucleares)11,15,19 dentro de las 24 h posteriores a la lesión35 debido a las células endoteliales vasculares que regulan y expresan las moléculas de adhesión en sus membranas para ayudar a guiar a los neutrófilos al sitio de la lesión que eliminan intrusos microbianos, y aunque los restos de tejido también liberan enzimas líticas como citocinas, proteasas y radicales libres que empeoran la lesión de las neuronas, de la glía y de los vasos sanguíneos al activar otras células inflamatorias y gliales para cascadas inflamatorias que pueden provocar lesiones neuronales o la muerte.11,19 Segundo, se lleva a cabo un reclutamiento y migración de monocitos y macrófagos11,15 (aproximadamente 2-3 días) que se vuelven fenotípica y morfológicamente indistinguibles de la microglía,35 secretan numerosas citocinas, radicales libres y factores de crecimiento que les permiten fagocitar el tejido dañado11,19 y muerto (neuronofagia y astrofagia)15 buscando promover el crecimiento axonal14 y la regeneración neural;18 sin embargo, aunque estos linfocitos segreguen factores de crecimiento para la supervivencia de las neuronas y la reparación de tejidos; los radicales libres y las citocinas proinflamatorias que también liberan contribuyen a la expansión de la lesión, empeorando su impacto.19
La respuesta inflamatoria es crítica para la eliminación de los desechos celulares, lo que puede prevenir la regeneración de las neuronas supervivientes. Sin embargo, la sobreactivación de la respuesta inflamatoria puede dañar el tejido sano y exacerbar la lesión.19 El reclutamiento de las células inmunitarias en el SNC lesionado iniciada a las pocas horas después del trauma medular y mantenido por varios días está dirigido por múltiples familias de proteínas: la molécula de adhesión intercelular-1 (MAIC-1; que promueve la infiltración de los neutrófilos en los tejidos), la selectina p, las citoquinas como interleucinas (IL) 1, 611,15 y 10,19 el factor de necrosis tumoral alfa (TNF-α), las proteínas inflamatorias de macrófagos (MIP)-1α y ß11,15 y otros agentes quimiotácticos como las quimiocinas y sus receptores que contribuyen a la infiltración celular y a la lesión,18 los mediadores inflamatorios como bradiquinina, prostaglandinas, leucotrienos, factor activador de plaquetas y serotonina;21 y el factor nuclear Kappa-B (NF-κB) que es un factor de transcripción que activa una variedad de genes que regulan respuestas inflamatorias, proliferativas y de muerte celular;11 las cuales se acumulan en el sitio de la lesión. Las citocinas, el óxido nítrico, el oxígeno y la expresión de especies de nitrógeno adicionales se activan, acelerando la respuesta inflamatoria del SNC. 21
Debido a la destrucción de la BHE, la fuga de suero y plasma, el aumento de las reacciones inflamatorias y la activación de los procesos anteriormente mencionados en el sitio de lesión,36 se activan las células microgliales dependiendo de la severidad de la lesión y adquieren morfología ameboide, expresando moléculas del complejo mayor de histocompatibilidad clase I y II (MHC I y II), el receptor de la fracción C3 del complemento (CD3) y el marcador de la activación de macrófagos ED1 (nombre relacionado al gen humano EDA o “ectodisplasina A”); transformándolas en células fagocíticas y coadyuvando con los macrófagos y monocitos. Las células inflamatorias pueden persistir por semanas.15
5.2. Pérdida de mielina y del crecimiento axonal en la médula espinal
La respuesta inmunitaria bifásica activa, la muerte de oligodendrocitos desencadenada por la excitotoxicidad del glutamato y exacerbada por una cascada que incluye apoptosis, ataques de radicales libres, actividades de mediador proinflamatorio/inflamatorio y citosinas, lo que promueve el daño progresivo del tejido e inhibe la regeneración neural; los macrófagos y la microglía contribuyen a la lisis de los oligodendrocitos por un mecanismo que involucra la producción de TNF-α y óxido nítrico,11,19 a la muerte neuronal por necrosis y a la desmielinización de los axones sobrevivientes debido a la sensibilización del sistema inmune a algunos componentes de la mielina; iniciando en las primeras 8 a 24 horas después de la LME11,15 y alcanzando su punto máximo en los próximos días.18 Posteriores a 48 horas de la lesión, los fagocitos sanguíneos acumulados localmente eliminan también la mielina degenerada y otros residuos celulares.15 Con la pérdida de mielina, los axones están expuestos a los efectos dañinos de los ROS y las citocinas inflamatorias, lo que lleva a incrementar la pérdida neuronal por necrosis y/o apoptosis. La desmielinización también produce retrasos/bloqueos en la conducción nerviosa dado que los axones que atraviesan el sitio de la lesión son la única conexión remanente entre el cerebro y las neuronas espinales caudales. La pérdida de mielina es particularmente evidente en las fases subagudas y crónicas.19
Además de la pérdida de mielina, el daño medular a este nivel de tiempo ocasiona la liberación de diversas moléculas que inhiben el crecimiento axonal; por ejemplo, los oligodendrocitos dañados exponen un tipo de proteínas inhibitorias específicas denominadas Nogo (del inglés Neurite outgrowth inhibitor) que están relacionadas con la inhibición del crecimiento axonal. Aunque hay células gliales que son dañadas externamente por el choque espinal, los oligodendrocitos dañados que expresan Nogo sufrieron daño externa e internamente, debido a la ausencia de la membrana externa de la molécula inhibitoria. Otro de los componentes inhibitorios del crecimiento axonal derivados de la mielina es la glicoproteína asociada a mielina (MAG) que puede promover o inhibir el crecimiento de neuritas. Las proteínas como las colapsinas-semaforinas (colapsina 2) constituyen una gran familia de glucoproteínas transmembrana que tienen efecto repulsivo a la guía axonal. Las netrinas o las moléculas de la familia de la tenasina-ECM (janusina-restrictina) también ejercen una actividad inhibitoria del crecimiento neuronal.15 La disminución en la expresión de receptores AMPA-kainato ocasiona que los oligodendrocitos dañados no puedan mejorar la conducción de los impulsos nerviosos a través de las neuronas.32
En esta fase también se da otro proceso conocido como cromatólisis central, que ocurre después de que una lesión en la neurona es sostenida o irreparable. Se caracteriza por la inflamación del cuerpo celular y la desaparición de los cuerpos de Nissl de la porción central de la célula, acompañada por una reubicación periférica del núcleo. Es una reacción somática donde los cambios parecen reflejar modificaciones reversibles en el metabolismo celular como la isquemia, interpretados como un estado de mayor actividad metabólica que favorece la regeneración axonal; causando degeneración de las vainas de mielina (a nivel de SNC y SNP) y muerte neuronal (a nivel de SNC), agravando notablemente el daño y la disfunción neurológica después de la LME. La cromatolisis y la inflamación son características de las células neuronales con microcirculación comprometida.19
5.3. Alteraciones proteicas en la lesión de médula espinal
La expresión de ARNm para la proteína proteolipídica (PLP), la principal proteína estructural en la mielina del SNC expresada por oligodendrocitos, se ve disminuida a las 24 horas, así como la proteína ácida fibrilar glial (GFAP), filamento intermedio principal de los astrocitos maduros con la expresión relativamente específica en estas células. Una semana después, la expresión del ARNm de la PLP puede incrementar sutilmente quizá por la alta proliferación de células precursoras de oligodendrocitos; así como la GFAP, relacionándose con un proceso de gliosis. Otra proteína que se afecta tras esta patología es la nestina, que es un filamento intermedio que se encuentra en las células madre que rodean el canal central de la ME sana, contribuye con nuevas células precursoras gliales y ependimarias de reemplazo para su reparación, viéndose disminuida tras el daño, pero recuperando sus niveles de concentración aproximadamente 3 días después, permaneciendo más altas de lo normal durante semanas. Tras el proceso excitotóxico desencadenado y la degeneración membranal de las neuronas por el daño primario, se da una disminución profunda en la expresión de las proteínas del receptor de glutamato-2 (GluR2) y NR1 (proteínas de subunidades clave para los tipos de receptores de glutamato AMPA y NMDA, respectivamente, presentes en muchas neuronas de la ME) desde las 24 horas hasta los 3 días posteriores al daño.37
Generalmente, durante las primeras 24 horas posteriores a la LME permanecen algunas células nerviosas intactas en el sitio dañado, sin embargo, los procesos del daño primario desencadenan otra cascada de mecanismos bioquímicos que comprometen el funcionamiento adecuado de las neuronas y células gliales que hasta en ese tiempo habían quedado ilesas. A esta segunda cascada bioquímica se le conoce como daño secundario y conlleva a una muerte neuronal secundaria.
6.1. Apoptosis en la médula espinal
Durante la fase aguda, el trauma mecánico en la ME provoca muerte celular de forma instantánea por necrosis, un proceso de inflamación celular y ruptura de la membrana celular. En cuestión de horas, otro tipo de muerte celular asume el centro del escenario:19 se le conoce como muerte celular programada o apoptosis;11 donde las células no se inflaman antes de su muerte, en donde el material genético y organelos citoplásmicos se condensan y separan en pequeños fragmentos en una ruta programada que requiere síntesis de energía y proteínas. Este tipo de muerte surge a raíz de la activación de las citoquinas, las lesiones inflamatorias, los daños ocasionados por los radicales libres y la excitotoxicidad,11,19 y se observa después de 3 horas hasta las 8 semanas.21 La cascada apoptótica se inicia en neuronas ocasionando pérdida de células, en oligodendrocitos contribuyendo a la desmielinización durante las primeras semanas después de la LME, en microglía contribuyendo a la lesión inflamatoria y, quizá, en astrocitos. Esta apoptosis ocurre a través de vías extrínsecas e intrínsecas mediadas por la activación de caspasas.11,18
El inicio de la apoptosis (principalmente en neuronas) se da por la vía intrínseca18,29 o independiente de receptor, que se activa por señales intracelulares como lo son las altas concentraciones de Ca++ intracelular11 que altera el potencial de membrana interno mitocondrial,18,19 abriendo el poro de transición/daño mitocondrial y liberando al citocromo C que se acopla con el factor inductor de apoptosis-1 (AIF-1) para activar la caspasa 9 (que activa a las caspasas efectoras 3 y 6), para culminar en la muerte de la neurona afectada. Por su parte, la vía extrínseca o dependiente de receptor se activa por diversas señales extracelulares, donde la más importante es la acumulación del TNF-α, que se acumula rápidamente en la ME lesionada y activa al receptor Fas de las neuronas, de la microglía y de los oligodendrocitos, induciendo una secuencia programada de activación de caspasas que involucra la caspasa 8 como inductora y las caspasas 3 y 6 como efectoras. La activación de las caspasas efectoras lleva a la muerte a las células afectadas. Un inductor alternativo de la vía extrínseca es la iNOS inducible, producida por los macrófagos, que también provoca la activación de caspasa-3 para efectuar la muerte celular programada.11,18 Así mismo, las calpaínas junto con las caspasas de ambas vías descomponen las proteínas en el citoesqueleto interno y la membrana de la célula, conduciendo a la muerte celular apoptótica.19
6.2. Daño mitocondrial y efecto de las ciclooxogenasas en la médula espinal
La LME altera la capacidad de las mitocondrias para llevar a cabo la respiración celular y la fosforilación oxidativa, modificando aún más la absorción de Ca++ dependiente de la respiración al inhibir el transporte mitocondrial de Ca++ y, por lo tanto, perturbando la homeostasis intracelular de este ion, alterando la permeabilidad de la membrana interna mitocondrial, incrementando la excitotoxicidad, contribuyendo a la inflamación osmótica y a la lisis mitocondrial, y generando más muerte celular y neuronal (principalmente por apoptosis).18
Aunque en el daño primario la COX-1 se encuentra activa, ésta sigue permitiendo la entrada de ácido araquidónico de la membrana celular rota al lugar con actividad enzimática, contribuyendo a la reducción del flujo sanguíneo por agregación plaquetaria, a la vasoconstricción y a la respuesta inflamatoria y peroxidación lipídica, donde ésta última entra en un ciclo repetitivo que involucra la producción de radicales libres.11,38 Estos radicales continúan dañando las membranas, con mayor peroxidación lipídica y formación de más radicales libres. La segunda isoforma de la COX, la COX-2, no se encuentra presente normalmente en la célula, pero la expresión de la proteína y del ARNm de ésta aparece rápidamente tras la exposición a lipopolisacáridos o citocinas proinflamatorias desencadenados por la cascada inflamatoria18,38 o por el aumento de neurotransmisores excitadores a nivel extracelular, generando una muerte neuronal por toxicidad directa;11 ello representa un sustrato común que une el daño de la membrana y la excitotoxicidad en la LME. La activación de las fosfolipasas asociadas a la membrana debido a la entrada de Ca++ y la liberación de ácido araquidónico promueve el aumento de los neurotransmisores excitadores extracelulares y evoca la activación neuronal, dando como resultado la inducción de la expresión de COX-2 en las neuronas corticales, que a su vez, resulta en muerte neuronal por toxicidad directa.18
6.3. Área de cavitación y cicatriz glial en la médula espinal
La desmielinización y la ruptura de la glía producen necrosis. Por su parte, la hemorragia y la isquemia local promueven la reactivación de las células gliales11,15 que, eventualmente, experimentan cambios morfológicos y funcionales como: hipertrofia, extensión de sus procesos y producción de más citocinas proinflamatorias (en un proceso conocido como “gliosis reactiva, glía activada o glía reactiva”).39,40 Esto propicia la conformación de las zonas de penumbra isquémica (durante la primera semana después del trauma) en las que tendrá lugar la muerte neuronal secundaria, donde se formarán cavidades y quistes que son discernibles en la sustancia gris y blanca. Los quistes están formados principalmente por glía reactiva, que involucra a la microglía y los astrocitos, así como a poblaciones de células periféricas en el área de la lesión.15,18 Por su parte, las áreas de cavitación se ven relacionadas a la isquemia, a la hemorragia, a la actividad lisosómica y/o a la infiltración e inflamación de macrófagos, conduciendo al abandono de los procesos neuronales por parte de los astrocitos. El receptor de manosa de macrófagos (MMr) y el receptor del complemento tipo 3 (Mac-1) integrina β2 están implicados en la cascada que induce la formación de estas cavidades.17,19
En un intento del SN por aislarse del resto del organismo, la superficie de los astrocitos reactivos cambia, incrementando su expresión de filamentos intermedios que son reconocidos por anticuerpos contra la GFAP (convirtiéndose en astrocitos fibrosos). A la par, existe un aumento en la concentración del factor de crecimiento fibroblástico básico (FGF-2) que promueve la proliferación de fibroblastos y angiogénesis permitiendo así que la capa recién formada de estos astrocitos fibrosos sea cubierta por material de matriz extracelular como laminina, fibronectina y colágeno de diferentes tipos, de fibroblastos del tejido conjuntivo adyacente, células de Schwann y macrófagos;15 formando así una verdadera barrera proteica física y química conocida como cicatriz glial, que sirve para el restablecimiento de la homeostasis iónica y de la integridad de la BHE pero que separa las neuronas que antes de la lesión estaban conectadas e inhibe la regeneración axonal.14,15 La conversión de los astrocitos a su modo fibroso se debe a la expresión de proteoglicanos inhibidores de la iniciación, adhesión, crecimiento y orientación de las neuritas. Esta respuesta alcanza un máximo a los 14 días de la lesión, pero permanece hasta 28 días después.15 Aunque la glía reactiva y la cicatriz glial forman una verdadera barrera para el restablecimiento de nuevas conexiones, también promueve el restablecimiento de la homeostasis iónica y de la integridad de la BHE11,14 al permitir que las neuronas que han perdido su inervación original sean inervadas por neuronas cercanas no dañadas. Cabe mencionar que este restablecimiento no conduce a la recuperación de la función original.15
Siendo ya la glía reactiva y habiéndose dañado las células dendríticas, la microglía residente y las células mononucleares reclutadas facilitan la exposición y la presentación de antígenos en el contexto del MHC clase II, que junto con sustancias co-estimuladoras como las citocinas proinflamatorias (Th1 [IL-2, IFNγ] y Th2), continúan favoreciendo la activación y expansión del repertorio autorreactivo de los timocitos CD4+ que mantienen el proceso autoinmune.15
6.4. Zona de penumbra y degeneración walleriana en la médula espinal
El proceso degenerativo de la ME lesionada continúa y se extiende en el tiempo y en el espacio. La llamada muerte neuronal secundaria o tardía comienza uno o dos días después de la lesión y es responsable de la muerte de más neuronas que las que ocasiona la muerte neuronal primaria. El tejido neural cercano al área lesionada o conectado con ella, presenta actividad eléctrica y actividad funcional deprimidas, llamando a esta área “zona de penumbra”; característica principal del daño secundario de la ME. Debido a que las neuronas muertas no son reemplazadas, los axones lesionados no se regeneran y los trastornos funcionales son permanentes.15 La pérdida de mielina es dependiente del tiempo, por lo que a los 7 días ya se presentan los axones sin protección de mielina y la desmielinización se incrementa después de 2 semanas debido al efecto constante de las células inflamatorias que entran en una segunda fase de migración.11 Los segmentos de axones separados por acción del flujo anormal de Ca++ y todos sus procesos desencadenantes permanecen intactos durante días después de la LME, pudiendo transmitir potenciales de acción cuando se estimulan. Finalmente, los axones se hinchan y se produce una desintegración granular catastrófica del citoesqueleto; la cual se completa en 1 hora. Los restos de mielina y axón en el SNP son eliminados por los macrófagos circulantes y células de Schwann; mientras que en el SNC se eliminan principalmente por la microglía activada y, de forma limitada, por los astrocitos.16 Sin embargo, éstos principalmente proliferan para rellenar los espacios una vez ocupados por las fibras nerviosas y así dar paso a la re-mielinización.18
En las zonas adyacentes a la lesión comienza a un proceso de re-mielinización debido a varios mecanismos: disminución de macrófagos, presencia y maduración de formas inmaduras de oligodendrocitos no dañados y liberación de factores de crecimiento de tipo neurotrófico como neurotrofinas por parte de las células de Schwann (las cuales pertenecen al SNP, pero se infiltran al sitio de lesión después del daño en médula espinal).15 La depleción/desaparición de macrófagos fagocíticos, a las semanas o meses, en las zonas adyacentes a la lesión medular deja una cavidad llamada quiste, carente de células y llena de fluido cerebroespinal, y rodeado de glía reactiva glía reactiva.15,41 Por su parte, el proceso de crecimiento y re-mielinización en algunos axones, así como pérdida del diámetro axonal, denominado degeneración walleriana,15,42 ocurre aproximadamente de los 14 días a las tres semanas en humanos y primates, instaurándose aproximadamente a las 3 semanas; y alrededor de las 24-48 horas en ratas jóvenes.15,16 Las células de Schwann regulan negativamente la expresión de genes asociados con mielina (Egr2/Krox20, MPZ, MBP, MAG y periaxina) y comienzan a proliferar y a aumentar las moléculas características de su etapa inmadura (L1, NCAM, p75NTR y GFAP); además, no sólo revierten su fenotipo a una etapa de desarrollo anterior, sino que se someten a una fase adecuada de "transdiferenciación" en la que adquieren características únicas, convirtiéndose en verdaderas "células de reparación".42 También algunas formas inmaduras de oligodendrocitos no dañados presentes maduran y son capaces de re-mielinizar algunos axones. Sin embargo, aunque los axones tengan el medio para volver a crecer, su recuperación funcional en humanos a menudo es incompleta debido a que la respuesta regenerativa de la neurona lesionada y de las células que rodean el axón de la neurona lesionada no puede mantener una respuesta efectiva que promueva el crecimiento durante largos períodos.41
6.5. Estabilización de la lesión medular en la médula espinal
La degeneración walleriana ya iniciada sigue conduciendo a la eliminación de los desechos inhibitorios en el nervio periférico y a la producción de un entorno que apoya el crecimiento de axones (aproximadamente a los 6 meses).41 Dos años después de la LME, ésta ha madurado y estabilizado completamente. La formación de la cicatriz glial sigue en proceso y la degeneración walleriana de los axones severamente lesionados se ha instaurado. Sin embargo, se puede producir una disfunción neurológica tardía, como la formación de quistes y siringes.14
La mayoría de las investigaciones en el campo de la lesión de médula espinal se enfocan en la preservación, restauración y activación de los sistemas motores descendentes, así como de los circuitos locales intraespinales que promuevan la recuperación de la función motora. Estas investigaciones han utilizado terapias mediante factores de crecimiento o neurotróficos (como la glía envolvente olfatoria, el factor de crecimiento neural, el factor de crecimiento derivado del cerebro, el factor de crecimiento básico de los fibroblastos, la neurotrofina-3, entre otros); mediante trasplantes (como de las células de Schwann, de las células envolventes del bulbo olfatorio, de las células progenitoras multipotenciales, de oligodendrocitos, astrocitos y fibroblastos, entre otros); mediante tratamientos farmacológicos (como la metilprednisolona, la naloxona, la nimodipina, el riluzol, entre otros); electrofisiológicos y alternativos (como la electroacupuntura y la fisioterapia). Sin embargo, a la fecha actual aún no hay tratamientos efectivos disponibles que reduzcan el daño primario y secundario que suceden después de la LME o que promuevan la recuperación de la función de esta importante estructura. La falta de intervenciones exitosas se debe a que no hay un entendimiento completo de los cambios bioquímicos, fisiológicos y neuroanatómicos que ocurren en diferentes tiempos después de la lesión espinal. Como se describió en esta revisión, la LME comprende un gran número de procesos bioquímicos relacionados con el daño primario y secundario involucrados en distintos tiempos y que aún se continúan investigando en la actualidad para poder obtener una recuperación. La importancia de poder comprender el orden de aparición y sus posibles desencadenantes de este complejo bioquímico ha permitido buscar un blanco terapéutico en el que se pueda reducir o eliminar algunos de estos procesos bioquímicos y, como consecuencia, disminuir la posibilidad de que los sujetos con lesión espinal pierdan completamente sus funciones motoras, sensoriales y autonómicas. Aún queda mucho más por investigar sobre los procesos bioquímicos producidos después de una lesión de médula espinal, no hay que perder de vista que la ciencia crece a pasos agigantados y que, en un posible futuro corto o a mediano plazo, se tenga un mapeo más completo que plasme todos los procesos bioquímicos involucrados en una lesión de médula espinal. Esto servirá para encontrar técnicas adecuadas que puedan inducir una restauración funcional más exitosa.
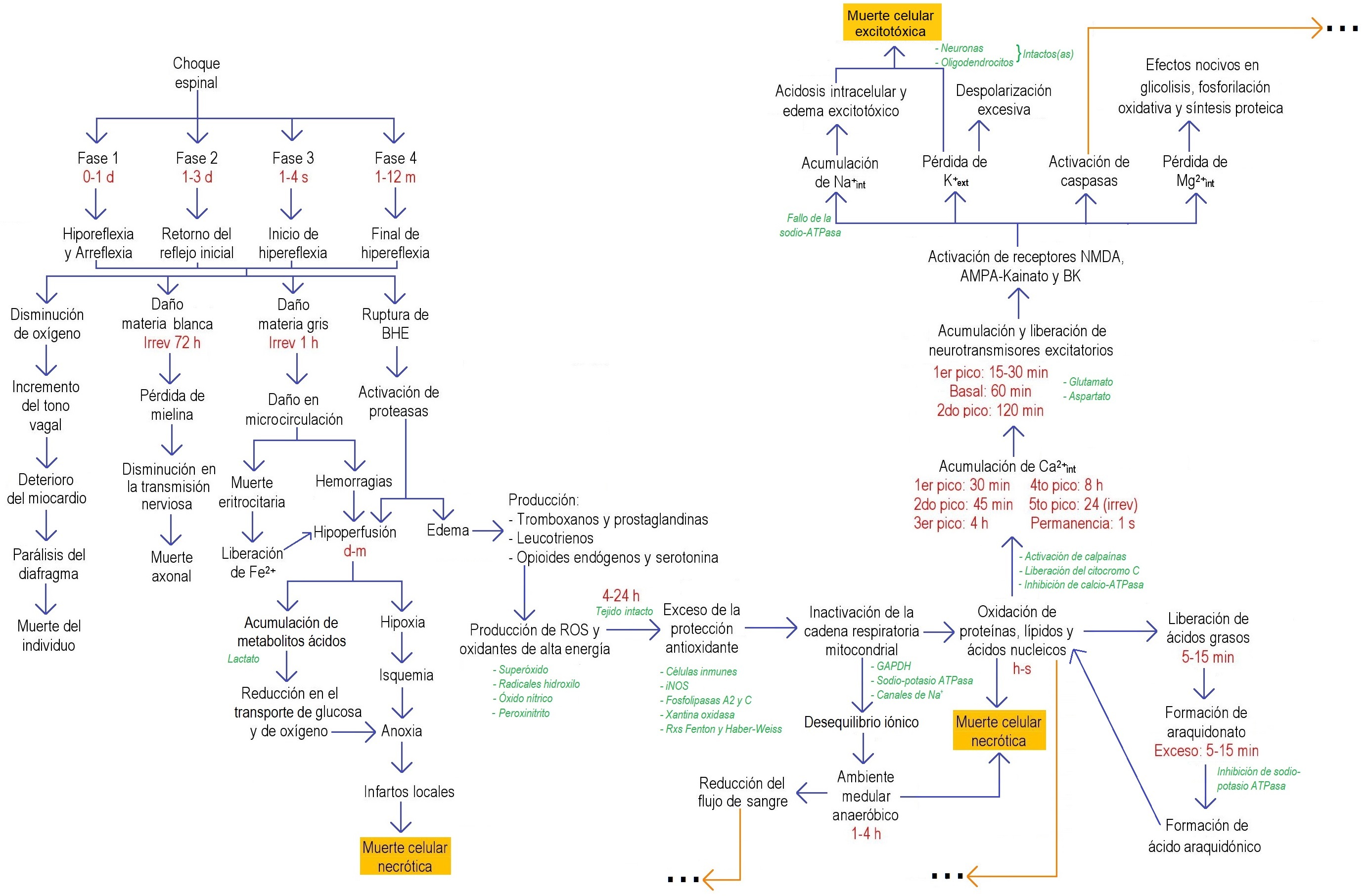
Figura 1. Diagrama de flujo de los procesos bioquímicos después de la LME en la fase aguda. Se muestra un bosquejo general de los procesos bioquímicos desencadenados tras una LME en la fase aguda. Estos mecanismos no siguen siempre el mismo orden y no siempre están presentes; dependen del sitio y del tipo de lesión espinal. AMPA: α-amino-3-hidroxi-5-metilisoxazol-4-propionato; ATP: trifosfato de adenosina; BHE: barrera hematoencefálica; d: día(s); ext: extracelular; f.c.: fase crónica; Fe+: ion fierro; f.s.a.: fase subaguda; GAPDH: gliceraldehido-3-fosfato deshidrogenasa; h: hora(s); iNOS: óxido nítrico sintasas; int: intracelular; min: minuto(s); NMDA: N-metil-D-aspartato; ROS: radicales libres o especies reactivas de oxígeno; rxs: reacciones; sem: semana(s); ◊▼ relacionan la fase aguda con la subaguda; ♠ relaciona la fase aguda con la crónica; ♦ relaciona la fase 2 con el proceso que desencadena en la fase aguda; ▲● relacionan la fase 3 y 4, respetivamente, con el proceso que desencadena en la fase crónica. Información tomada de: 11,12,28,29,31–36,38,39,13,40–46,15–17,20,22,26,27.
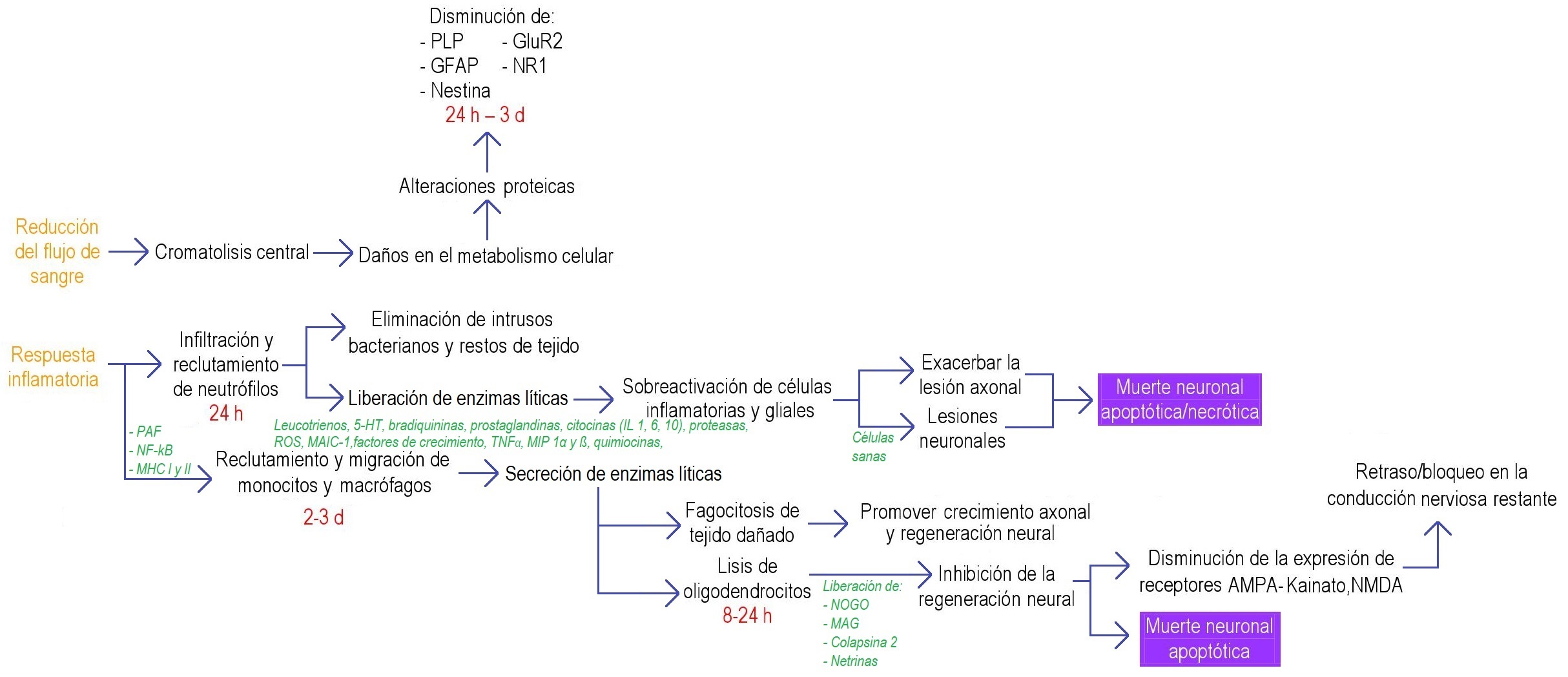
Figura 2. Diagrama de flujo de los procesos bioquímicos después de la LME en la fase subaguda. Se muestra un bosquejo general de los procesos bioquímicos desencadenados tras una LME en la fase subaguda. Estos mecanismos no siguen siempre el mismo orden y no siempre están presentes; dependen del sitio y del tipo de lesión espinal. 5-HT: serotonina; AMPA: α-amino-3-hidroxi-5-metilisoxazol-4-propionato; CR3: receptor de la fracción C3 del complemento; d: día(s); ED1: marcador de la activación de macrófagos; f.s.a.: fase subaguda; GFAP: proteína ácida fibrilar glial; GluR2: receptor de glutamato-2; h: hora(s); IL: interleucina; MAG: glicoproteína asociada a mielina; MAIC-1 molécula de adhesión intercelular-1; MHC: complejo mayor de histocompatibilidad; MIP: proteínas inflamatorias de macrófagos; NF-kB: factor nuclear Kappa-B; NMDA: N-metil-D-aspartato; NOGO: inhibidor del crecimiento de neuritas (neurite outgrowth inhibitor); NR: receptor de NMDA; PAF: factor activador de plaquetas; PLP: proteína proteolípida; ROS: radicales libres o especies reactivas de oxígeno; TNF-α: factor de necrosis tumoral alfa; ◊▼ relacionan la fase aguda con la subaguda; ♦ relaciona la fase 2 del choque espinal (fase aguda) con el proceso que desencadena en la fase subaguda. Información tomada de: 11,12,28,29,31–36,38,39,13,40–46,15–17,20,22,26,27
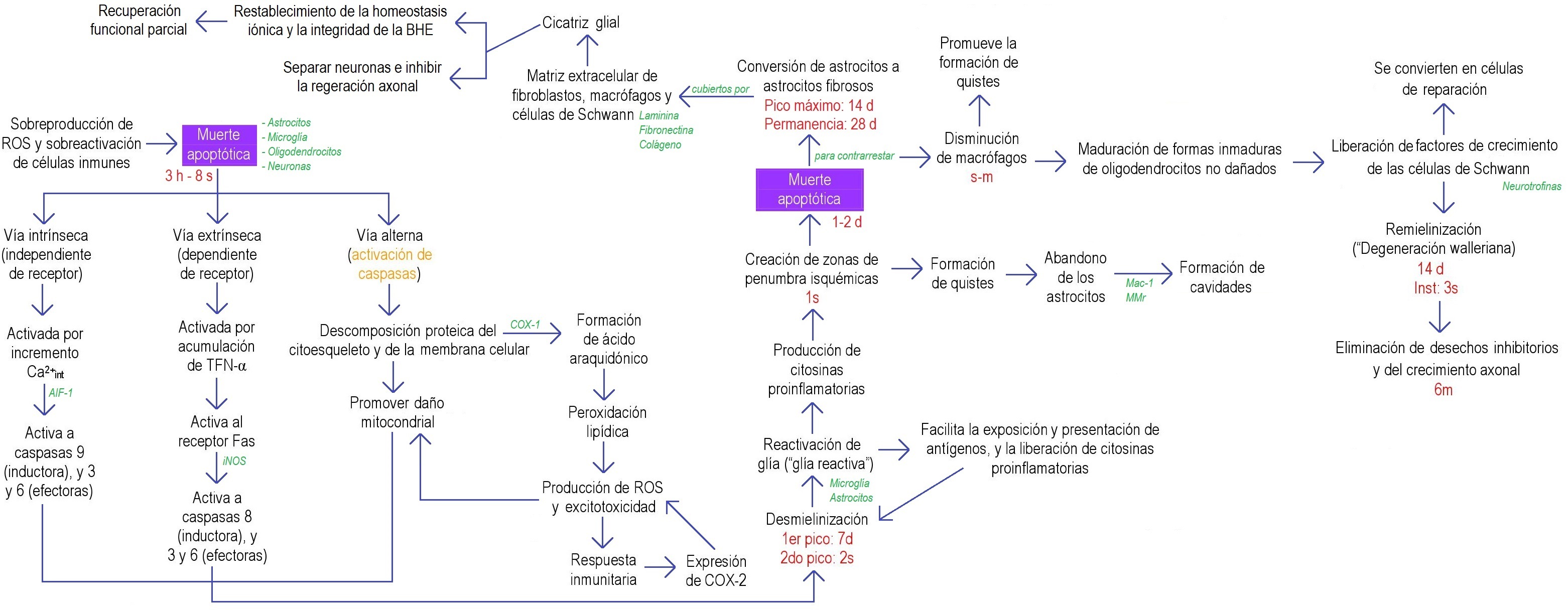
Figura 3. Diagrama de flujo de los procesos bioquímicos después de la LME en la fase crónica. Se muestra un bosquejo general de los procesos bioquímicos desencadenados tras una LME en la fase crónica. Estos mecanismos no siguen siempre el mismo orden y no siempre están presentes; dependen del sitio y del tipo de lesión espinal. AIF-1: factor inductor de apoptosis-1; BHE: barrera hematoencefálica; COX: ciclooxigenasas; d: día(s); f.c.: fase crónica; h: hora(s); iNOS: óxido nítrico sintasas; inst: instaurar; int: intracelular; Mac-1: receptor del complemento tipo 3; MMr: receptor de manosa de macrófagos; ROS: radicales libres o especies reactivas de oxígeno; sem: semana(s); TNF-α: factor de necrosis tumoral alfa; ♠ relaciona la fase aguda con la crónica; ▲● relacionan la fase 3 y 4 del choque espinal (fase aguda), respetivamente, con el proceso que desencadena en la fase crónica. Información tomada de: 11,12,28,29,31–36,38,39,13,40–46,15–17,20,22,26,27
Los autores de este artículo de revisión han apoyado en su contenido y en la elaboración del formato de este escrito. Todos los autores declaran que no tienen conflicto de interés alguno en su publicación.
Se agradece al Consejo Nacional de Ciencia y Tecnología (CONACyT) por la beca otorgada a S.Y.L.A. (número 893213).
1. Kaye A, Davis S. Principles of neurophysiological assessment, mapping and monitoring. Springer; 2014. 34,36,37.
2. Velayos J, Diéguez G. Anatomía y fisiología del sistema nervioso central. CEU; 2015. 35, 37, 38, 61 p.
3. Moreno S. Trauma raquimedular. Morfolia. 2015;7(1):48–54.
4. SWorld Health Organization, International Spinal Cord Society. International Perspectives on Spinal Cord Injury. WHO. 2014;1–250.
5. Pérez R, Martín S, Renán S, Ortiz S. Aspectos epidemiológicos de la lesión medular de la población del Centro Nacional de Rehabilitación. Rev Mex Med Física y Rehabil. 2008;20:74–82.
6. Pérez C, Sánchez D, López M, Morgado C, Beltran L, Coria G, Hernández M, Manzo J. Aplicaciones terapéuticas para la lesión de médula espinal. Neurobiol Rev electrócnica. 2018;9(21):1–16.
7. National Spinal Cord Injury Statistical Center. Spinal Cord Injury. Facts and Figures at a Glance. NSCISC. 2015;3.
8. Estrada S, Carreón A, Parra M, Ibarra C, Velasquillo C, Vacanti C. Lesión de médula espinal y medicina regenerativa. Rev Salud Pública México. 2007;49.
9. Mancilla A, García G. Frecuencia de la lesión medular pediátrica en un centro de rehabilitación. Experiencia de nueve años. Rev Mex Neurocienc. 2018;4:36–44.
10. Organización Mundial de la Salud. Lesiones medulares [Internet]. OMS. 2013. Available from: http://www.who.int/es/news-room/fact-sheets/detail/spinal-cord-injury
11. Acevedo J, Varón L, Berbeo M, Lee O, Díaz R. Avances fisiopatológicos para el entendimiento de la lesión medular traumática . Revisión bibliográfica. Rev Colomb Ortop y Traumatol Av. 2008;22:272–281.
12. Alizadeh A, Dick S, Abdolrezaee S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front Neurol. 2019;10(282):1–25.
13. Allbutt H, Siddall P, Keay K. Contusive spinal cord injury evokes localized changes in NADPH-d activity but extensive changes in Fos-like immunoreactivity in the rat. J Anat. 2007;211(3):352–370.
14. Ballesteros V, Marré B, Celmira M, José F, Juan Z. Lesión de la médula espinal. Actualización bibliográfica: Fisiopatología y tratamiento inicial. Coluna/ Columna. 2012;11(1):73–76.
15. Caballero S, Nieto M. Fisiopatología de la lesión medular. Revisión de literatura. Rev Vet México. 2005;36(1):75–86.
16. Shi F, Zhu H, Yang S, Liu Y, Feng Y, Shi J, Xu D, Wu W, You S, Ma Z, Zou J, Lu P, Xu XM. Glial response and myelin clearance in areas of wallerian degeneration after spinal cord hemisection. J Neurotrauma. 2009;26:2083–2096.
17. 17. Denecke C, Aljovic A, Bareyre F. Combining molecular intervention with in vivo imaging to untangle mechanisms of axon pathology and outgrowth following spinal cord injury. Exp Neurol. 2019;318:1–11.
18. Dumont R, Okonkwo D, Verma S, Hurlbert J, Boulos P, Ellegala D, Dumont A. Acute spinal cord injury , part I : pathophysiologic mechanisms. Clin Neuropharmacol. 2001;24(5):254–264.
19. Oyinbo C. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp. 2011;71:281–299.
20. Papa S, Mauri E, Rossi F, Perale G, Veglianese P. Introduction to spinal cord injury as clinical pathology. In: Spinal Cord Injury (SCI) Repair Strategies. Elsevier; 2020. p. 1–12.
21. Yilmaz T, Turan Y, Keleş A. Pathophysiology of the spinal cord injury. J Clin Exp Investig. 2014;5(1):131–136.
22. Pérez CA, Concha A, Hernández ME, Manzo J. Influence of the paraventricular nucleus and oxytocin on the retrograde stain of pubococcygeus muscle motoneurons in male rats. Brain Res. 2005. 1041:11–18.
23. Xu Y, Zheng Z, Ping K, Qian Z. Effects of spinal cord injury on c-fos expression in hypothalamic paraventricular nucleus and supraoptic nucleus in rats. Brain Res. 2006;1087:175–179.
24. Del-Bel E, Borges C, Delfino H, Guimaräes F. Induction of Fos protein immunoreactivity by spinal cord contusion. Brazilian J Med Biol Res. 2000;33:521–528.
25. Chang E. Expresión de c-Fos en el cerebelo después de la lesión espinal de la rata macho. Universidad Veracruzana. 2013. p. 1-85.
26. Visavadiya N, Springer J. Altered Cerebellar Circuitry following Thoracic Spinal Cord Injury in Adult Rats. Neural Plast. 2016;2016:1–5.
27. Ditunno JF, Little JW, Tessler A, Burns AS. Spinal shock revisited: A four-phase model. Spinal Cord. 2004;42(7):383–395.
28. Villa J, Villamar D, Toscano J, Blancas L, Herrera J, Silva R. Current developments in antioxidant therapies for spinal cord injury. In: Spinal Cord Injury Therapy. 2019. p. 1–32.
29. Díaz-Ruiz A, Méndez-Armenta M, Nava-Ruiz C, Garduño C, Santander I, Ruiz A, Ríos C. Fenobarbital administrado sólo o en combinación con dapsona inhibe apoptosis y mejora la función motora después de una lesión traumática de la médula espinal en ratas. Arch Neurociencias. 2015;20(4):240–246.
30. Delgado A, Minguillón C, Joglar J. Fármacos moduladores de los metabolitos del ácido araquidónico. In: Introducción a la química terapéutica. 2nd ed. Madrid: Díaz de Santos; 2004. p. 399.
31. Tan BL, Norhaizan ME, Liew WPP, Rahman HS. Antioxidant and oxidative stress: A mutual interplay in age-related diseases. Front Pharmacol. 2018;9(1162):1–28.
32. Mcdonald JW, Sadowsky C. Spinal-cord injury. Lancet. 2002;359:417–425.
33. Jacobsen M, Lett K, Barden J, Simpson GL, Buttigieg J. Activation of the large-conductance, voltage, and Ca2+ - activated K+ ( BK ) channel in acute spinal cord injury in the wistar rat is neuroprotective. Front Neurol. 2018;9(1107):1–12.
34. Perrin F, Najib H. Serotonergic mechanisms in spinal cord injury. Exp Neurol. 2019;318:174–191.
35. Gensel JC, Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015;1619:1–11.
36. Nathan FM, Li S. Environmental cues determine the fate of astrocytes after spinal cord injury. Neural Regen Res. 2018;12(12):1964–1970.
37. Wu X, Yoo S, Wrathall JR. Real-time quantitative PCR analysis of temporal-spatial alterations in gene expression after spinal cord contusion. J Neurochem. 2005;93(4):943–952.
38. García J, Gómez J. Fisiopatología de la ciclooxigenasa-1 y ciclooxigenasa-2. Elsevier. 2000;27(1):33–35.
39. Okada S, Hara M, Kobayakawa K, Matsumoto Y, Nakashima Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci Res. 2018;126:39–43.
40. Pérez Capote K. Respuesta de las células gliales al daño neuronal in vitro. Universidad de Barcelona. Universidad de Barcelona; 2006. p. 1-56.
41. Gaudet AD, Popovich PG, Ramer MS. Wallerian degeneration: Gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. 2011;8(110):1–13.
42. Pellegatta M, Taveggia C. The complex work of proteases and secretases in wallerian degeneration: Beyond neuregulin-1. Front Cell Neurosci. 2019;13(93):1–13.
43. Witiw CD, Fehlings MG. Acute Spinal Cord Injury. J Spinal Disord Tech. 2015;28(6):202–210.
44. Fariña M, De la Barrera S, Marqués M, Velasco F, Vázquez G. Update on traumatic acute spinal cord injury. Part 2. Elsevier. 2017;41(5):306–315.
| Recibido: 05 de agosto, 2020 | Aceptado: 11 de noviembre, 2020 |
*Correspondencia en: Dr. César Antonio Pérez-Estudillo. Dirección: Centro de Investigaciones Cerebrales, Universidad Veracruzana. Médicos y Odontólogos s/n, Col. Unidad del Bosque, C.P. 91010, Xalapa, Veracruz, México. Teléfono: +52 (228) 8418900 Ext. 16304, E-mail: cesperez@uv.mx.mx
Este es un artículo de libre acceso distribuido bajo los términos de la licencia de Creative Commons, (http://creamasal@unam.mxtivecommons.org/licenses/by-nc/3.0), que permite el uso no comercial, distribución y reproducción en algún medio, siempre que la obra original sea debidamente citada.