Neurodegenerative diseases: The spinocerebellar ataxia type 7 in Mexico
- Top
- Editorial Board
- Guide for Authors
- Recommend to Friend
- Contact the Editor
- Legal
- About Us
- Indexing
- ISSN 2007-3054
- Centro de Investigaciones Cerebrales
Universidad Veracruzana
Review Article
Enfermedades neurodegenerativas: La ataxia espinocerebelosa tipo 7 en México
Rolando Garcia-Martinez1*, Elizabeth Hernandez2, Rebeca Toledo2**, Oscar H. Hernandez1*, Maria Elena Hernandez2**, Luis I. Garcia2***, Shirley Ortiz3, Jorge Manzo2***
1Centro de Investigaciones Biomedicas, Universidad Autonoma de Campeche, Mexico. 2Centro de Investigaciones Cerebrales, Universidad Veracruzana, Xalapa, Ver., Mexico. 3Instituto de Rehabilitacion para las Ataxias en Mexico, A.C., Mexico. *Cuerpo Academico de Biomedicina, Universidad Autonoma de Campeche. **Cuerpo Academico de Neuroquimica, Universidad Veracruzana. ***Cuerpo Academico de Neurociencias, Universidad Veracruzana.
Abstract/Resumen
Introducción
Conclusiones
Agradecimientos
Referencias
Correspondencia
The spinocerebellar ataxias (SCAs) are a group of neurodegenerative diseases that have a genetic origin. Some are caused by a mutation in a gene that lead to the production of an abnormal protein called ataxin, a transcription factor that tends to form inclusions in the nucleus and cytoplasm of the cell. This alteration has been associated with the clinical and pathological manifestations of this disease. However, little is known about these diseases in many Latin American countries. Objective: The purpose of this review is to present the current state of research on SCAs, its classification, and to describe a Mexican family diagnosed with SCA type 7 (SCA7), to understand its history and genealogy. Method: Searches of the PUBMED databases and files of the Instituto de Rehabilitacion para las Ataxias en Mexico (IRAM) were performed. Conclusion: Because it is important to describe the prevalence and frequencies of the SCAs in other states of Mexico, it is necessary to support research in this area, especially in government health institutions.
Key words: Spinocerebellar ataxia, Neurodegenerative disease, Triplet repeats, Ataxin.
Las ataxias espinocerebelosas (AECs) son un grupo de enfermedades neurodegenerativas que tienen un origen genético. Algunas son causadas por la mutación en un gen que conduce a la producción de una proteína anormal llamada ataxina, un factor de transcripción que tiende a formar inclusiones en el núcleo y el citoplasma de la célula. Esta alteración se ha asociado con las manifestaciones clínicas y patológicas de la enfermedad. Sin embargo, poco se sabe acerca de estas enfermedades en muchos países de América Latina. Objetivo. El propósito de esta revisión es presentar el estado actual de la investigación sobre las AECs, su clasificación y describir a una familia Mexicana diagnosticada con AEC tipo 7 (AEC7), para entender su historia y su genealogía. Método: La investigación se realizó de la base de datos del PUBMED y de los archivos del Instituto de Rehabilitación para las Ataxias en México (IRAM). Conclusión. Debido a la importancia de describir la prevalencia y la frecuencia de las AECs en otros estados de México, es necesario apoyar la investigación en esta área, especialmente en las instituciones de salud del gobierno.
Palabras clave: Ataxia espinocerebelosa, Enfermedad neurodegenerativa, Tripletes repetidos, Ataxina.
Neurodegenerative diseases represent a large group of disorders of the central nervous system (CNS). While the clinical and neuropathological characteristics of the various neurodegenerative diseases differ, they share the symptoms of neuronal degeneration, and the subsequent functional impairment of the affected areas.1
Two well-known and common forms of these diseases are Alzheimer’s2 and Parkinson’s diseases.3 Other, less common disorders include Huntington’s disease,4 multiple sclerosis,5 amyotrophic lateral sclerosis,6 Rett, Fragile X and Angelman syndromes,7 and SCAs.8 SCAs, which are addressed in this article, are a group of neurodegenerative disorders characterized by progressive ataxia that results from the degeneration of the cerebellum and other structures, and from the loss of their input and output connections to information.9,10
Ataxia derives from the Greek word "a-taxis", which means "without order". It is also used to describe a gait disorder, "drunk walk", which is characterized by instability, lack of coordination, and increased base of support.11,12 Ataxia results from the dysfunction of the cerebellum, spinal cord, and peripheral nerves, or a combination of the three conditions.10
Because most cerebellar degenerative disorders are genetically transmitted, they are also called "inherited ataxias”.13 Similar to other human neurodegenerative diseases of unknown etiology, their classification has been based on clinical and pathological criteria. However, these characteristics can vary even among members of the same family. As such, the classification needs to be determined by biochemical and molecular procedures that are focused on the involved genes.13
The purpose of this review is to present the current state of research on the SCAs and its classification, and to describe a Mexican family diagnosed with the SCA7, to understand its history and genealogy.
2. Inherited ataxias
This group includes neurodegenerative disorders characterized by a slowly evolving degeneration of cerebellar neurons and other different neural structures, including the spinal cord and basal ganglia.14 Many SCAs are caused by a mutation of the SCA gene, which produces an aberrant transcript of ribonucleic acid. When this transcript is translated into protein, there are repetitions of the corresponding amino acid, and the mutated protein tends to aggregate within nuclear inclusions.15,16 This phenomenon has been associated with the clinical and pathological manifestations of this disease.17
2.1 Classification
The pioneering work of Harding in the early 1980s initiated the clinical-genetic classification of this disorder, leading to the more recent classification based on molecular genetics.18-20 So, the current classification is based on the distinction between inherited and acquired variants, and each group has subclasses. The inherited ataxias are classified according to the specific genetic deficit, including autosomal dominant, autosomal recessive, mitochondrial diseases and X-linked ataxias. For most ataxia cases, it is possible to characterize the molecular genetic defect that causes the disease.11 The genetic defect or mutation may involve the substitution of a single base (adenine, guanine, cytosine or thymine) for another, the loss of any part of the sequence of one or more genes, or a trinucleotide, pentanucleotide or hexanucleotide repeat of these bases.
2.1.1 Autosomal dominant cerebellar ataxias (ADCA)
In ADCA (Figure 1), the gene causing the disease is found on a non-sex chromosome and, accordingly, affects women and men equally. When one parent (the father) has a recessive normal allele (n) and a dominant defective allele (D), and the other parent (the mother) has two normal alleles (nn), each of their sons has a 50% chance of inheriting the abnormal gene from the father and will have the same defect as the father.21
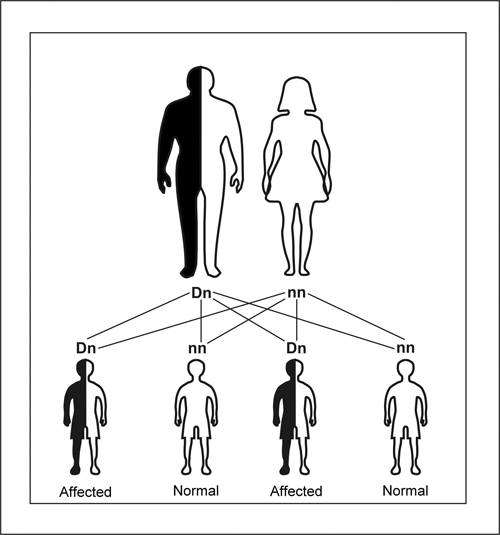
Figure 1. Schematic illustrating autosomal dominant inheritance. The affected parent has a defective allele (D), which dominates its normal counterpart (n). Each child has 50 a % chance of inheriting the D allele from the affected parent.
Different pathogenic mechanisms for autosomal dominant disorders have been identified. The disorders include the SCA type 1 (SCA1) to SCA36,8 with the exception of SCA6 (Table 1). Seven are caused by the expansion of CAG triplet repeats, with the exceptions of SCA10 and SCA31, which result from pentanucleotide repeats,10,22 and SCA36, which results from hexanucleotide repeats.23 SCA9 does not exist, but its name has been retained. The name SCA24 was assigned to the single recessive form of spinocerebellar ataxia,10 and dentatorubral-pallidoluysian atrophy (DRPLA) is included in this group.
All available evidence suggests that these disorders are caused by the abnormal function of a protein called "ataxin" (e.g., ataxin-1 for SCA1, ataxin-2 for SCA2, ataxin-7 for SCA7), which has a polyglutamine repeat sequence. In another group of dominant disorders, including episodic ataxias 1 to 7 (EA1-7)8 and SCA6 (Table 1), the mutations affect genes that encode ion channels.24
2.1.2 Autosomal recessive cerebellar ataxias (ARCA)
Diseases with an autosomal recessive inheritance pattern are generally rare, and their inheritance follows the expected Mendelian ratio of 3:1. Unlike dominantly inherited diseases, diseases with autosomal recessive inheritance require two copies of the defective gene for a person suffer the symptoms of the disease (Figure 2).
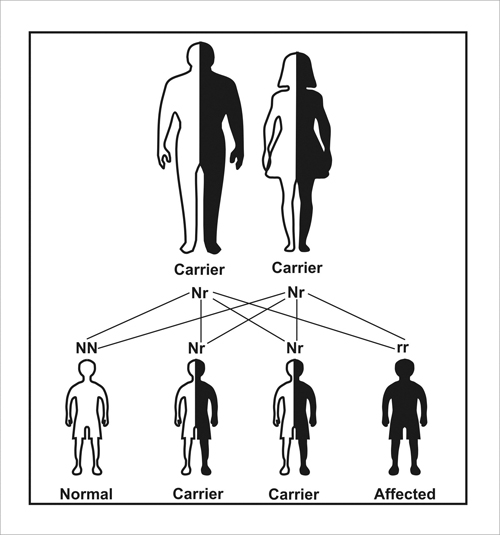
Figure 2. Schematic illustrating autosomal recessive inheritance. Both parents who usually do not have the disease, carry a normal dominant allele (N), which takes precedence over its defective and recessive counterpart (r). Each child has a 25% chance of inheriting two r alleles (leading to serious birth defects), a 25% chance of inheriting two N alleles (a normal phenotype), and a 50% chance of being a Nr carrier (as are both parents).
The autosomal recessive ataxias (Table 2) are caused by the loss of a mitochondrial protein, frataxin, which has been linked to respiratory function and iron homeostasis.25 Friedreich’s ataxia is, the most common type of recessive ataxia,13,26 and one of every 20,000 people in the United States suffers from Friedreich’s ataxia.8 In recessive disorders, the symptoms usually begin in childhood or in adolescence, rather than in adulthood. However, for reasons that remain unclear, the symptoms are not necessarily present at birth or during infancy.11 In the United States, the incidence of both ADCA and ARCA is 1-5 cases per 100,000 peoples.8
Table 1. List of autosomal dominant cerebellar ataxias and their mutations.8,27
Autosomal dominant cerebellar ataxia |
Due to |
SCA1 |
The expansion of CAG repeat on chromosome 6p21.3 |
SCA2 |
The expansion of the CAG repeat on chromosome 12q23-24.1 |
SCA3 |
The expansion of the CAG repeat on chromosome 14q32.1 |
SCA4 |
A mutation on chromosome 16q22.1 |
SCA5 |
A mutation located on chromosome 11p12-q12. Ataxia described in the ancestors of U.S. President Abraham Lincoln. |
SCA6 |
The expansion of the CAG repeat on chromosome 19p13 |
SCA7 |
The expansion of CAG trinucleotide on chromosome 3p12-21.1 (Associated with retinal lesions) |
SCA8 |
The expansion of triplet CTA / CTG on chromosome 13q21 |
SCA10 |
The expansion of the pentanucleotide ATTCT on chromosome 22q13 |
SCA11 |
A mutation on chromosome 15q14-21.3 |
SCA12 |
The expansion of the CAG repeat on chromosome 5q31-33 |
SCA13 |
A mutation on chromosome 19q13.3-13.4 |
SCA14 |
A mutation on chromosome 19q13.4 |
SCA15 |
A mutation on chromosome 3p24.2-3 |
SCA16 |
A mutation on chromosome 8q23-24.1 |
SCA17 |
The expansion of the CAG repeat on chromosome 6q27 |
SCA18 |
A mutation on chromosome 7q22-32 |
SCA19 |
A mutation on chromosome 1p21-q21 |
SCA20 |
A mutation on chromosome 11p13-q11 |
SCA21 |
A mutation on chromosome 7p21.3-15.1 |
SCA22 |
A mutation on chromosome 1p21-q23 |
SCA23 |
A mutation on chromosome 20p13-12.3 |
SCA25 |
A mutation on chromosome 2p15-21 |
SCA26 |
A mutation on chromosome 19p13.3 |
SCA27 |
A mutation on chromosome 13q34 |
SCA28 |
A mutation on chromosome 18p11.22-q11.2 |
SCA29 |
A mutation on chromosome 3p26 |
SCA30 |
A mutation on chromosome 4q34.3-q35.1 |
SCA31 |
A mutation on chromosome 16q22.1 |
SCA32 |
A mutation on chromosome 7 |
SCA33-35 |
Unknown |
SCA36 |
Mutation on chromosome 20p13 |
DRPLA |
Atrophy in the connections of the dentate-red-pallido nuclei. |
Episodic |
|
EA1 |
The mutation affect voltage-gated potassium channels function (KCNA1) |
EA2 |
The mutation affect the P/Q (CACNA1) and L-type (CACNB4) calcium channel |
EA3 |
A mutation on chromosome 1q42 |
EA4-6 |
Unknown |
EA7 |
A mutation on chromosome 19q13 |
Table 2. List of autosomal recessive cerebellar ataxias and their defects.8,27
Autosomal recessive cerebellar ataxias |
Associated with |
Friedreich´s ataxia |
GAA triplet expansions STM7 gene on chromosome 9q13-21 |
Abetalipoproteinemia or Bassen-Kornzweig syndrome |
Defects in the gene for the protein of microsomal triglyceride transfer of chromosome 2p24 |
Ataxia of Charlevoix-Saguenay |
Defects in the SACS gene, maps to chromosome 13q11 |
Ataxia with vitamin E deficiency |
Defects in a gene located on chromosome 8q13 |
Ataxia with oculomotor apraxia type 1 |
A mutation on chromosome 9p13 (apraxin) |
Ataxia with oculomotor apraxia type 2 |
A mutation on chromosome 9q34 (senataxin) |
Ataxia telangiectasia or Louis-Bar syndrome |
Defects in a gene located on chromosome 11q22-23 |
Boucher Neuhauser syndrome |
Retinopathy and hypogonadism |
Marinesco-Sjögren syndrome |
Defect in SIL1 gene for chaperone protein (HSPA5) |
Refsum disease |
Defect in PHYH gene placed on chromosome 10pter-p11.2 |
2.1.3 Mitochondrial diseases
Mitochondrial diseases are due to a mutation in the mitochondrial genes that are responsible for energy production. The characteristic symptom of these mitochondrial disorders is ataxic gait, and is often associated with other complications such as peripheral neuropathy, ophthalmoparesis, retinitis pigmentosa, etc. (Table 3).27
2.1.4 X-linked ataxias
X-linked ataxia is a disorder that affects men in one or more generations in the maternal line, and this ataxia is among the most common disorders observed (Table 3). The symptoms that occur most frequently include: ataxic gait, kinetic tremors, parkinsonism and polyneurophaty.27 Cognitive dysfunctions have also been observed.
3. Acquired ataxias
This group includes sporadic or acquired ataxias, which may be caused by chronic alcoholism, toxins and drugs (phenytoin, lithium, valproate, amiodarone, metronidazole, procainamide, mefloquine, isoniazida, metals and solvents), hypothyroidism, stroke, infectious diseases, and neoplastic disorders.27
This group also includes some degenerative, adult onset ataxias, such as multiple system atrophy (MSA), idiopathic late-onset cerebellar ataxia (ILOCA), sporadic cerebellar degeneration (SCD), striatonigral degeneration (SDC) and Shy Drager Syndrome (SDY).8
4. CAG repeats
Approximately 15 years ago, it was discovered that many neurodegenerative diseases are attributable to increases in unstable triplet repeats in DNA. To date, more than 35 SCAs have been described, and, in at least seven of these diseases, the repeated element is a CAG triplet coding for glutamine. Many diseases have been described to result from the formation of polyglutamine repeats. Other neurodegenerative diseases such as Alzheimer’s, amyotrophic lateral sclerosis, Parkinson’s disease, spinobulbar atrophy, and DRPLA atrophy, have been shown to involve the accumulation of mutated proteins in the nucleus and cytoplasm of the cell, and in the extracellular space.28
Table 3. List of mitochondrial diseases and X-linked ataxias.27
Mitochondrial diseases |
Due to |
Kearns-Sayre syndrome |
Deletions (loss of genetic material) in the mtDNA (mitochondrial deoxyribonucleic acid) |
MERRF syndrome (myoclonus epilepsy with ragged red fibers) |
Mutations in the mtDNA |
MELAS syndrome (mitochondrial encephalomypathy, lactic acidosis with stroke-like episodes) |
Mutations in the mtDNA |
NARP syndrome (neurophaty, ataxia, and retinitis pigmentosa) |
Mutations in the mtDNA ATPase gene |
|
|
X-linked ataxias |
|
FXTAS syndrome (Fragile-X tremor ataxia syndrome) |
A mutation in CGG triplet on the X chromosome |
Rett syndrome |
Defect in the MeCP2 gene on the X chromosome |
X-ALD (X-linked adrenoleukodystrophy) |
A mutation in the ABCD1 gene located on the X-chromosome |
Other congenital ataxias |
|
Multiple proteins contain areas of polyglutamine residues (polyQ) that are prone to instability and expansion. Up to a certain length, the occurrence of polyQ in the ataxin protein is not pathogenic. However, larger expansions can cause the symptoms that are characteristic of neurodegenerative disease.11
In the case of ataxin-7, 4 to 17 CAG repeats are considered to be in the normal range, with 10 being the most frequently observed number of repeats.15 The presence of greater than 37 repeats, (which is equivalent to 37 glutamine amino acids), is associated with the clinical and pathological features of this disease.19
5.Treatment of the SCAs
In general, treatments for neurodegenerative diseases are lacking, and therapeutic interventions, mostly comprise symptomatic and palliative measures. Neurodegenerative diseases constitute a terrible disability, and can cause physical and psychological suffering in patients and their families.1
Currently, there is no cure for spinocerebellar ataxias, and preclinical and clinical studies with insulin-like growth factor-I (IGF-I) are ongoing. The serum levels of IGF-I are altered in animal models of ataxia and human patients,29 but relationship between these altered levels and disease pathology is unclear. Nevertheless, this relationship may be a target for the pharmacological treatment of ataxia.30
The Centro para la Investigacion y la Rehabilitacion de las Ataxias Hereditarias (CIRAH) in Holguin, Cuba, has also conducted important research on the effects of zinc sulfate supplementation in SCA2 patients.10 Zinc sulfate supplementation was found to be associated with improvements in coordination, gait, balance and cognitive functions, including attention, concentration, and memory, as well as executive function. However, the mechanism by which these effects are mediated is unknown. The CIRAH has plans to open a department of neuropathology, which will potentially be one of the continent’s largest centers for the development of this area of research.
6. SCAs in Mexico
The existence of this disease in Mexico was first reported by Matsuura et al., in 2000. They found an expansion of a pentanucleotide (ATTCT) repeat in intron 9 of SCA10 gene in all patients examined, who belonged to five Mexican families. SCA10 is an autosomal dominant disorder characterized by cerebellar ataxia and seizures.31
In 2001, Rasmussen et al., reported 18 cases of SCA10 in four Mexican families. They described the molecular findings in these patients, and reported an expansion of ATTCT repeats ranging from 920 to 4140 repeats, with an average age at onset of 26.7 years. The clinical signs were more significant, and included pan cerebellar ataxia and seizures. Pyramidal signs, ocular dyskinesia, cognitive impairment and/or behavioral disturbances were also observed. Thus, SCA10 may affect tissues other than the cerebellum.32
In 2007, Alonso et al., reported 108 dominant families and 123 sporadic cases in the Mexican Mestizo population. The distribution of ADCA was 45.4% for SCA2, 13.9% for SCA10, 12% for SCA3, 7.4% for SCA7 and 1.6% for SCA17. They identified six individuals with the rare allele (CAG) 33, and two with early onset ataxia. These results showed the existence of different SCA in Mexico, and suggested the need for designing testing strategies for the general Mexican population.33
In other countries, however, research in the field of spinocerebellar ataxias has been ongoing for decades. The reason for the delay in Mexico may be the lack of knowledge of the clinical and pathological features of the disease. The family physicians or physical therapists who frequently examine people with any type of motor disorder may be unaware that they are observing a case of spinocerebellar ataxia. Additionally, patients may be dying of other complications without having been diagnosed with SCA.
The Instituto de Rehabilitacion para las Ataxias en Mexico (IRAM) is located in Xalapa, the capital of the state of Veracruz.El IRAM es una asociación civil que surge como un intento por atender casos específicos de ataxias hereditarias, a los cuales no se les brindaba el seguimiento adecuado en ningún hospital de Gobierno. Asimismo, la participación del Centro para la Investigación y Rehabilitación de las Ataxias Hereditarias de Cuba (CIRAH), ha sido fundamental para asesorar y capacitar al personal del IRAM en este campo, de manera que puedan detectar familias xalapeñas y de municipios aledaños afectadas por ataxias espinocerebelosas, que anteriormente habían pasado desapercibidas. Ahora, ambas instituciones trabajan en coordinación para brindar apoyo a las personas afectadas por esta enfermedad. The IRAM is a civil association that was founded in 2006 by a family with a number of suspected cases of ataxias and whom adequate management was not provided by any hospital in the state. The Instituto Nacional de Neurologia y Neurocirugia (INNN) “Dr. Manuel Velasco Suarez” in Mexico City is currently the only public hospital that offers presymptomatic tests and genetic testing for individuals who arrive from other states of Mexico. However, much remains to be done by the Institutes and Health Center in terms of the detection and referral of individuals who have the classic symptoms of ataxia. No records exist in other hospitals for hereditary ataxias, and there is a lack of institutions specializing in the monitoring and care of patients with these neurodegenerative diseases.
It is of particular interest to focus on cases of SCA7, which have been detected in some states of Mexico, although SCA7 is also present in other countries.34-39
6. Spinocerebellar ataxia type 7
SCA7 is an autosomal dominant cerebellar ataxia that is associated with progressive macular degeneration. It was formerly known as olivopontocerebellar atrophy type III13 and is now known as spinocerebellar ataxia type 7.39,40
Many families around the world and from different ethnic groups have been reported to have SCA7. These locations include North (USA and Mexico) and South America (Brazil and Peru), Europe (Belgium, Finland, France, Germany, Spain, Sweden and United Kingdom), Africa (Algeria, Morocco, Libya, Tunisia and South Africa), and Asia (China, Japan, Korea and Philippines).20,35,39,41-45
The population of Mexico largely derives from the Spanish who came to Mexico during the “Conquest” (XVI century) and the slaves brought mainly from Africa, who settled in this territory in communities and later merged with the local people. This population diversity might account for the distribution of some the SCAs, such as the SCA7, in the Mexican Mestizo population.
6.1.1 Clinical Features
SCA7 is characterized by progressive cerebellar ataxia; ophthalmoplegia; dysarthria; dysphagia; decreased movements saccades and visual acuity; pyramidal and extrapyramidal signs; deep sensory loss; and in some cases, symptoms of dementia.19,41,46 Most patients with SCA7 suffer from progressive macular dystrophy and, in advanced stages of the illness, permanent blindness.17 Some patients have difficulty with controlling their sphincters, increased reflexes in the lower extremities, and decreased vibration perception.15 As with other SCAs,47 olfactory deficits have also been reported in patients with SCA7.48 SCA7 evolves through four clinical states, and “the electroretinogram is a potential biomarker of disease progression”.49
6.1.2 Neuropathological features
The neuropathological features that have been reported to accompany SCA7 include a moderate to severe loss of neurons (PkC and granule cells) and gliosis in the cerebellum,46 inferior olive, dentate nuclei, pontine nuclei and structures related to the motor system such as globus pallidus, substantia nigra, subthalamic nuclei, red nuclei and spinal cord.36,46 The loss of bipolar cells, ganglion cells and photoreceptors in the retina have also been observed,42 as have neuronal loss and gliosis of the lateral geniculate body and occipital cortex.17
6.1.3 Genetic Characteristics
The mutation responsible for SCA7 is the expansion of a CAG triplet repeat in the first exon of the SCA7 gene, which is located on chromosome 3p12-p21.1.15,16,18,36,43,46,50-53 The gene encodes a protein of unknown function, called ataxin-7, which is made up of 892 amino acids and has a polyglutamine domain in the amino terminal region.15,46,53 The mutated form of ataxin-7 is restricted to the neuronal nucleus, where it tends to form insoluble aggregates within nuclear inclusions.16,19,54 This clustering has been associated with the clinical and pathologic features of the disease.17,45,55 The presence for more than 37 repeats is associated with the disease,19 and more than 200 repeats have been observed.10
Genetic anticipation is often observed in SCA7, as is the case for the rest of the autosomal dominant cerebellar ataxias and in other diseases produced by CAG repeats. Specifically, the number of repeats present is inversely proportional to the age of onset of symptoms and to the intensity of clinical involvement.44,55
6.2. Cases of SCA7 in Mexico
In 2004, Rolon-Lacarriere et al., reported a case of a Mexican family (Durango state) of 13 individuals from three generations who had ataxia and other neurological disorders. The clinical history of the individual was obtained, and a neurological exam, including neuropsychological studies, neurophysiologic, ophthalmologic, neuroradiologic, and genetic tests was conducted.45 A genealogical study revealed that the family had a history of vision and gait disturbances. In all members of the last generation, the observed symptoms included global cerebellar syndrome, pyramidal, visual impairment and varying degrees of ophthalmoparesis, maculopathy with progressive retinal degeneration, and atrophy of the cerebellum, brainstem and the cerebral hemispheres. Clinical anticipation was observed in three subjects, in whom the symptoms were more severe, and onset was earlier in the youngest generation. Molecular diagnosis was only performed in the father, and PCR analysis showed 46 CAG repeats in the SCA7 gene region. Clinical and genetic studies showed that the disease was spinocerebellar ataxia type 7, and this was the first publication of SCA7 in a Mexican family.45
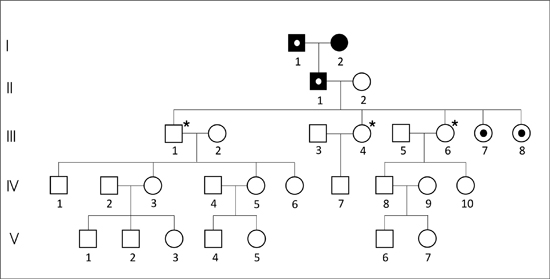
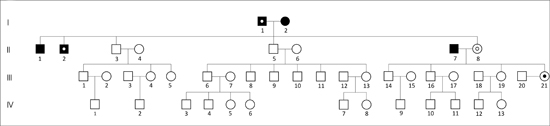
Here, we present the case of a family in Xalapa, which comprises 126 peoples across five generations. A genealogical tree was constructed and was divided into three subfamilies (Figures 3 A, B, C) for clarity. The man in the first generation in the three subfamilies is the same and is thougth to be the index carrier of the mutation. Subfamily A is composed of 29 individuals distributed into five generations, including 14 males (48.3%) and 15 females (51.7%). Subfamily B is composed of 43 individuals distributed into four generations, with 26 males (60.4%) and 17 females (39.6%). The subfamily C is composed of 54 individuals, distributed into five generations, including 27 males (50%) and 27 females (50%). According to the data provided by family members and the IRAM files, 12 individuals might have been affected, of whom five had already died. Of the seven remaining members, only three women of ages 38 (subfamily A-III,8), 39 (subfamily B-III,21), and 41 (subfamily A-III,7), were confirmed to have SCA7 by the INNN.
These women had the characteristic expansion of triplets on chromosome 3p that was associated with retinal lesions, leading to diagnosis of spinocerebellar ataxia type 7. This was the first report of SCA7 in the state of Veracruz.
In 2006 da Cunha et al., reported a case series of SCA7 in individuals from five consanguineous marriages in Brazil, suggesting that the incidence of the disease increases in closely related individuals.56 In the case presented above, no consanguineous marriages were reported. One member of subfamily B (II, 2) and one of the subfamily C (IV,20) who were suspected to have the disease were not married and had no offspring. Similarly, the three women with genetic confirmation of SCA7 had no children, which contributed to the low incidence of cases in these subfamilies.
However, the number of states in Mexico with cases of SCAs could rise if the proper diagnosis of the disease is applied widely. This would lead to a more accurate estimation of the frequency of the disease, improve the availability of appropriate management and provide better quality of life for people affected by this devastating condition.
7. Future research in SCA7
Helmlinger et al., (2004) showed that ataxin-7, the protein that arises from the mutated of SCA7 gene, is a subunit of TFTC (TATA-binding protein-free TAF-containing complex) and STAGA (SPT3/TAF9/GCN5 acetyltransferase complex).57 These transcriptional complexes are linked to histone acetylation,58 both in nucleosomes and in the free protein form. Moreover, severe transcriptional alterations have been detected in several cellular models, including yeast and mouse cells that had glutamine repeats in the ataxin-7 protein. These date suggest that this abnormality interferes with the normal function of the transcription factors, which are mislocalized to form nuclear inclusions.59 However, the mechanism that facilitates transcriptional deregulation and protein accumulation remains unknown.
The mouse retina is a suitable model for studying transcriptional mechanisms. Specialized photoreceptor cells of the retina, rods and cones express specific genes coding for components of the phototransduction cascade, the process involved in converting light signals to electrical signals. Rods represent over 70% of all retinal cells. Helmlinger et al., (2004) showed that in the retina of SCA7 mutant mice, the TFTC/STAGA complex induces the hyperacetylation of histone H3.57 As a result, the cores of the rods show chromatin decondensation, which correlates with the severe transcriptional repression of target genes.58 As such, the retinal degeneration observed in the mouse model of SCA7 may be mediated by the loss of expression of genes that are necessary for phototransduction.59,60
More recently, Schaefer et al., (2012) suggested a role for the polyQ sequences in some proteins, including ataxin. They mentioned that the expanded polyQ sequences facilitate their interaction with other proteins by stabilizing the complex, which is necessary for the transcription process. The abnormal amount of polyQ alters this interaction, resulting in the formation of protein aggregates in inclusion bodies.61
Subfamily A
Subfamily B
Subfamily C
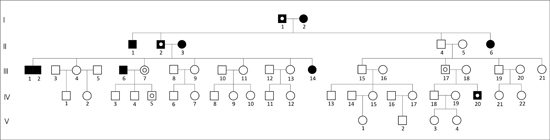
Figure 3. Pedigree of three Mexican subfamilies (A, B, C). The squares or black circles indicate the deceased. The black point inside the circles indicates persons whose genomic DNA was analyzed by PCR in the INNN, and were confirmed to have SCA7. The asterisk indicates a lack of SCA7. The white point inside the squares or circles indicates individuals with suspected disease. The man of the first generation in the three subfamilies is the same.
Young et al., (2007) reported that ataxin-7 is a substrate for caspases and possibly other proteases, and is degraded to form cytotoxic fragments. They showed that ataxin-7 is cleaved by caspase-7 at the Asp sites at position 266 and 344 and that the proteolytic processing of ataxin-7 may contribute to the pathogenesis of SCA7.62<
Several proteins are responsible for cell proteostasis, including chaperones that promote folding and decrease proteotoxicity. The accumulation of misfolded proteins can lead to a loss of function and cell death. It has been shown in cell culture and the mouse brain that the overexpression of chaperone proteins prevents aggregation and decreases polyglutamine toxicity.63 These findings may lead to identification of suitable targets for the development of therapies.
The spinocerebellar ataxias are genetic diseases that can pass from one generation to another, without a family knowing that one or both of the parents carry the disease gene. This lack of knowledge ocurrs because most of the SCA are manifested in adulthood (after 40 years of age), after most people have already reproduced. Moreover, research into drugs to alleviate symptoms of the disease does not show signs of success in the near future, although in vivo animal experiments focused on IGF-I, caspases and chaperones, and the supplementation of ataxia patients with zinc sulfate hold some promise.
Therefore, it is essential to gain awareness of the existence of such diseases, though their incidence in our country is low, so that the family physicians in charge of primary care have the knowledge to detect these diseases and properly refer patients to an appropriate institution for follow up.
We wish to acknowledge Marlene, Ivonne and Maria Luisa of IRAM, for their contributions to this work.
- Saenz de Pipaon I, Larumbe R. Programa de Enfermedades Neurodegenerativas. An Sist Sanit Navar 2001 24 (suppl 3): 49-76.
- Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid ß peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatr Neurosci 2012 37: 1-18.
- Wu T, Hallett M. The cerebellum in Parkinson´s disease. Brain 2013 136: 696-709.
- Tasset I, Sanchez F, Tunez I. Bases moleculares de la enfermedad de Huntington: papel del estres oxidativo. Rev Neurologia 2009 49: 417-424.
- Rio-Izquierdo J, Montalban X. Natalizumab en esclerosis multiple. Rev Neurologia 2009 49: 265-269.
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld H, Shoesmith C, Strong J, Woolley SC. Practice Parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, and respiratory therapies (an evidence-based review). Neurology2009 73: 1218-1226.
- Pabello M. La proteina MeCP2 y sus implicaciones en enfermedades neurodegenerativas. Degree Thesis. Xalapa Ver, Mexico; 2007.
- Fogel BL. Ataxia Classification. National Ataxia Foundation 2013: 1-2.
- Klockgether T, Evert B. Genes involved in hereditary ataxias. Trends Neurosci 1998 21: 413-418.
- Velazquez L. Ataxia espinocerebelosa tipo 2. Principales aspectos neurofisiologicos en el diagnostico, pronostico y evolucion de la enfermedad. Holguin (Ed.), La Habana. 2006 pp 5-113.
- Nance M. Living with ataxia. 2nd (Ed.), New York. 2003 pp 2-39.
- Ganong WF. Fisiologia Medica. 10th (Ed.), Mexico,2002 pp 239-244.
- Plaitakis A. Classification and epidemiology of cerebellar degenerations. In: Plaitakis A (Ed.). Cerebellar Degenerations: Clinical Neurobiology. Massachusetts: Kluwer Academic Publishers. 1992 pp 185-204.
- Stevanin G, Lebre AS, Zander C, Cancel G, Durr A, Brice A. Autosomal dominant cerebellar ataxia with progressive pigmentary macular dystrophy. In: Manto MU, Pandolfo M (Ed.). The Cerebellum and its Disorders. Cambridge University Press. England 2002 pp 459-468.
- David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 1997 17: 65-70.
- Jonasson J, Juvonen V, Sistonen P, Ignatius J, Johansson D, Bjorck EJ, Wahlstrom J, Melberg A, Holmgren G, Forsgren L, Holmberg M. Evidence for a common Spinocerebellar ataxia type 7 (SCA7) founder mutation in Scandinavia. Eur J Hum Genet 2000 8: 918-922.
- Cancel G, Duyckaerts C, Holmberg M, Zander C, Yvert G, Lebre AS, Ruberg M, Faucheux B, Agid Y, Hirsch E, Brice A. Distribution of ataxin-7 in normal human brain and retina. Brain 2000 123: 2519-2530.
- Benomar A, Krols L, Stevanin G, Cancel G, LeGuern E, David G, Ouhabi H, Martin J, Durr A, Zaim A, Ravise N, Busque C, Penet C, Van Broeckhoven C, Brice A. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. Nat Genet 1995 10: 84-88.
- David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada M, Grid B, Holmberg M, Yahyaoui M, Hentati F, Chkill T, Agid Y, Brice A. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet 1998 7: 165-170.
- Kim Y, Park SS, Joo SI, Kim JM, Jeon BS. Molecular analysis of spinocerebellar ataxias in Koreans: frequencies and reference ranges of SCA1, SCA2, SCA3, SCA6 and SCA7. Mol Cel 2001 12: 336-341
- Papalia DE, Wendkos S. Desarrollo Humano. McGraw-Hill/Interamericana, Mexico. 1990 pp 52-57.
- Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, Ishino F, Saito Y, Murayama S, Yoshida M, Hashizume Y, Takahashi Y, Tsuji S, Shimizu N, Toda T, Ishikawa K, Mizusawa H. Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 2009 85: 544-557.
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat inNOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011 81: 121-130.
- Adelman JP, Bond CT, Pessia M, Maylie M. Episodic ataxia results from voltage-dependent potassium channels with altered function. Neuron 1995 15: 1449-1454.
- Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997 276: 1709-1712.
- Chevis CF, da Silva CB, D´Abreu A, Lopes-Cendes I, Cendes F, Bergo FP, Franca MC. Spinal cord atrophy correlates with disability in Friedreich´s Ataxia. Cerebellum 2013 12: 43-47.
- Marmolino D, Manto M. Past, present and future for cerebellar ataxias. Curr Neuropharmacol 2010 8: 41-61.
- Grisolia S. Genetica de las enfermedades neurodegenerativas. In: Segovia de Arana JM, Mora-Teruel F (Ed.). Enfermedades Neurodegenerativas. Serie Cientifica Farmaindustria. Madrid 2002 pp 131-157.
- Fernandez AM, Carro EM, Lopez-Lopez C, Torres-Aleman I. Insulin-like growth factor I treatment for cerebellar ataxia: Addressing a common pathway in the pathological cascade?. Brain Res Rev 2005 50: 134-141..
- Bitoun E, Finelli MJ, Oliver PL, Lee S, Davies KE. AF4 is a critical regulator of the IGF-1 signaling pathway during Purkinje cell development. J Neurosci 2009 29: 15366-15374.
- Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 2000 26: 191-194.
- Rasmussen A, Matsuura T, Ruano L, Yescas P, Ochoa A, Ashizawa T, Alonso E. Clinical and genetic analysis of four Mexican families with spinocerebellar ataxia type 10. Ann Neurol 2001 50: 234-239.
- Alonso E, Martinez-Ruano L, De Biase I, Mader C, Ochoa A, Yescas P, Gutierrez R, White M, Ruano L, Fragoso-Benitez M, Ashizawa T, Bidichandani SI, Rasmussen A. Distinct distribution of autosomal dominant spinocerebellar ataxia in the mexican population. Movement Disord 2007 22: 1050-1053.
- Velazquez-Perez L, Rodriguez-Labrada R, Canales-Ochoa N, Sanchez-Cruz G, Fernandez-Ruiz J, Medrano-Montero J, Aguilera-Rodriguez R, Diaz R, Almaguer-Mederos LE, Palomino-Truitz A. Progression markers of Spinocerebellar Ataxia 2. A twenty years neurophysiological follow up study. J Neurol Sci 2010 290: 22-26.
- Gouw LG, Digre KB, Harris CP, Ptacek LJ. Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology 1994 44: 1441-1447.
- Bird TD, Pagon RA, La Spada AR. Spinocerebellar Ataxia type 7. GeneReviews1998 1: 1-15.
- Martin JJ, Regemorter NV, Krols L, Brucher JM, Barsy T, Szliwowski H, Evrard P, Ceuterick C, Tassignon MJ, Dieleman HS, Delatte H, Willems PJ, Broeckhoven CV. On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol 1994 88: 227-286.
- Abe T, Tsuda T, Yoshida M, Wada Y, Kano T, Itoyama Y, Tamai M.(2000). Macular degeneration associated with aberrant expansion of trinucleotide repeat of the SCA 7 gen in 2 Japanese families. Arch Ophthalmol 2000 118: 1415-1421.
- Castañeda MA, Avalos C, Jeri FR. Clinical and genetic studies of a family from Peru affected by spinocerebellar ataxia type 7. Rev Neurol 2000 31: 923-928.
- Gouw LG, Castañeda MA, McKenna CK, Digre KB, Pulst SM, Perlman S, Lee MS, Gomez C, Fischbeck K, Gagnon D, Storey E, Bird T, Jeri FR, Ptacek LJ. Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum Mol Genet 1998 7: 525-532.
- Mayo D, Yusta A, Vazquez JM, Garcia-Ruiz P, Robledo M, Benitez J. Ataxia espinocerebelar tipo VII (AEC 7). Comunicacion de una familia española afectada. Rev Neurol 1999 28: 964-966.
- Holmberg M, Johansson J, Forsgren L, Heljbel J, Sandgren O, Holmgren G. Localization of autosomal dominant cerebellar ataxia associated with retinal degeneration and anticipation to chromosome 3p12-p21.1. Hum Mol Genet 1995 4: 1441-1445.
- Gu W, Wang Y, Liu X, Zhou B, Zhou Y, Wang G. Molecular and clinical study of spinocerebellar ataxia type 7 in Chinese kindreds. Arch Neurol 2000 57: 1513-1518.
- Johansson J, Forsgren L, Sandren O, Brice A, Holmgren G, Holmberg M. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet 1998 7: 171-176.
- Rolon-Lacarriere O, Rasmussen-Almaraz A, Hernandez-Cruz H, Carranza-del Rio J, Gonzalez-Cruz M, Gutierrez-Moctezuma J. Ataxia espinocerebelosa de tipo 7: descripcion de una familia mexicana. Rev Neurol 2004 38: 736-740.
- Holmberg M, Duyckaerts C, Dürr A, Cancel G, Gourfinkel-An I, Damier P, Faucheux B, Trottier Y, Hirsch E, Agid Y, Brice A. Spinocerebellar ataxia type 7 (SCA7): A neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet 1998 7: 913-918.
- Fernandez Ruiz J, Diaz R, Hall‐Haro C, Vergara P, Fiorentini A, Nuñez L, Drucker‐Colin R, Ochoa A, Yescas P, Rasmussen A, Alonso E. Olfactory dysfunction in hereditary ataxia and basal ganglia disorders. Neuroreport 2003 14: 1339-1341.
- Galvez-Zuñiga VH, Diaz-Perez R, Fernandez-Ruiz J. Identificacion olfativa en pacientes con ataxia espinocerebelar tipo 7. 11th Meeting of Grupo de Bioseñales. Xalapa, Ver., Mexico 2012.
- Horton LC, Frosch MP, Vangel MG, Weigel-DiFranco C, Berson EL, Schmahmann JD. Spinocerebellar ataxia Type 7: Clinical course, phenotype-genotype correlations, and neuropathology. Cerebellum 2013; 12: 176-193.
- Ström AL, Forsgren L, Holmberg M. Identification and characterization of spinocerebellar ataxia type 7 (SCA7) isoform SCA7b in mice. Biochim Biophys Acta 2005 1731: 149-153.
- Gatchel JR, Watase K, Thaller C, Carson JP, Jafar-Nejad P, Shaw C, Zu T, Orr HT, Zoghbi Y. The insulin-like growth factor patway is altered in spinocerebellar ataxia type 1 and type 7. P Natl Acad Sci USA 2008 105: 1291-1296.
- Chen S, Peng G, Wang X, Smith A, Grote S, Shoper B, Spada A. Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum Mol Genet 2004 13: 53-67.
- Garden G, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci 2002 15: 4897-4905.
- Latouche M, Fragner P, Martin E, Hachimi KH, Zander C, Sittler A, Ruberg M, Brice A, Stevanin G. Polyglutamine and polyalanine expansions in ataxin 7 result in different types of aggregation and levels of toxicity. Mol Cell Neurosci 2006 31: 438-445.
- Robitaille Y, Lopes-Cendes I, Becher M, Rouleau G, Clark A. The neuropathology of CAG repeat diseases: review and update of genetic and molecular features. Brain Pathol 1997 7: 901-926..
- da Cunha S, Goes W, Marques W. Spinocerebellar ataxia type 7 (SCA7). Family princeps, history, genealogy and geographical distribution. Arq Neuro-psiquiat 2006 64: 222-227.
- Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N, Van-Dorsselaer A, Wurtz JM, Mandel JL, Tora L, Devys D. Ataxin-7 is a subunit of GCN5 histone acetyltransferase containing complexes. Hum Mol Genet 2004 13: 1257-1265.
- McCulloug SD, Grant PA. Histone acetylation, acetyltransferasa, and ataxia-alteration of histone acetylation and chromatin dynamics is implicated in the pathogenesis of polyglutamine-expansion disorders. Adv Protein Chem Struct Biol 2010 79: 165-203.
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmüller A, Picaud S, Zoghbi H, Trottier Y, Tora L, Devys D. Glutamine-expanded Ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. Plos Biol 2006 4: 432-445.
- Helmlinger D, Abou-Sleymane G, Yvert G, Rousseau S, Weber C, Trottier Y, Mandel JL, Devys D. Disease progression despite early loss of polyglutamine protein expression in SCA7 mouse model. J Neurosci2004 24: 1881-1887.
- Schaefer MH, Wanker EE, Andrade-Navarro MA. Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res 2012 40: 4273-4287.
- Young JE, Gouw L, Propp S, Shoper BL, Taylor J, Lin A, Hermel E, Logvinova A, Chen SF, Chen S, Bredesen, DE, Truant R, Ptacek LJ, La Spada AR, Ellerby LM. Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcripcional dysregulation. J Biol Chem 2007 282: 30150-30160.
- Labbadia J, Novoselov SS, Bett JS, Weiss A, Paganetti P, Bates GP, Cheetham ME. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 2012 35: 1180-1196.
| Received: September 18, 2013 | Accepted: October 16, 2013 |
Correspondencia:
*Laboratorio de Neurofisiologia, Centro de Investigaciones Biomedicas, Universidad Autonoma de Campeche Av. Agustin Melgar s/n Col. Buenavista, Campeche, Camp. C.P. 24039 Tel. (981) 8119800 Ext. 2020100 Fax +52 981 813 01 76. email: rogarcia@uacam.mx.
This is an open access article distributed under the Creative Commons Attribution License (http://creativecommons.org/licenses/by-nc/3.0), which permits noncommercial use, distribution and reproduction in any medium, provided the original work is properly cited.